Notice and Comment – Draft Revised Excessive Price Guidelines (August 2008)
The Patented Medicine Prices Review Board is a quasi-judicial tribunal with the mandate to ensure that manufacturers´ prices of patented medicines sold in Canada are not excessive
Draft Revised Excessive Price Guidelines for comments by October 6, 2008
Table of Contents
- Introduction and Purpose of the Stakeholder Consultation
- Board Positions on Proposed Revisions to the Excessive Price Guidelines
- Preamble
- I - Legal Framework
- II - Policies
- III - Guidelines and Procedures
- Chapter 1 - The Scientific Review Process
- Chapter 2 - The Price Review Process
- Chapter 3 – Investigations
- Schedules
- Schedule 1 – Submissions by Patentees on Therapeutic Improvement
- Schedule 2 – Levels of Evidence for Determination of Therapeutic Improvement 3
- Schedule 3 – Comparable Dosage Forms
- Schedule 4 – Therapeutic Class Comparison Test
- Schedule 5 – Reasonable Relationship Test
- Schedule 6 – International Price Comparison Test
- Schedule 7 – International Therapeutic Class Comparison Test
- Schedule 8 – CPI-Adjustment and De-linking Methodology
- Schedule 9 – Criteria for Commencing an Investigation
Introduction and Purpose of the Stakeholder Consultation
The Patented Medicine Prices Review Board (PMPRB) has developed a Draft Revised Compendium of Policies, Guidelines and Procedures as part of the next phase of the Guidelines review. This draft document builds on the consultations launched in 2005 regarding the relevance and appropriateness of the Board´s Excessive Price Guidelines (Guidelines). This draft also builds on the advice of the various working groups established relative to specific issues including, for the Working Group on Price Tests, the potential impact of mandatory reporting of benefits which, following the Federal Court (FC) decision in the matter of LEO Pharma Inc. and the medicine Dovobet, was deferred to January 1, 2009 (i.e., for the January to June 2009 reporting period).
This consultation package is organized in two Parts: Part I summarizes the issues that have been under review, along with Stakeholders´ and Working Groups´ views, and Board Positions; and Part II is the Draft Revised Compendium of Policies, Guidelines and Procedures. Stakeholder comments are currently being requested on the Draft Revised Compendium. The Board will review all submissions and publish the final revised Compendium in mid-Fall. Your written feedback on this draft document should be sent directly to Ms. Sylvie Dupont, Secretary of the Board, no later than October 6, 2008, at the following address:
Box L40
Standard Life Centre
333 Laurier Avenue West
Suite 1400
Ottawa, Ontario, K1P 1C1
Or by email: sdupont@pmprb-cepmb.gc.ca
If your comments are significant in length, please include a summary highlighting the key points of your submission. As with previous consultations, all submissions received by the Board will be posted on its Web site as part of the PMPRB´s commitment to openness and transparency.
Board Positions on Proposed Revisions to the Excessive Price Guidelines
Over the past two years, extensive consultations have been held with PMPRB´s stakeholders in the form of bi-lateral and multi-lateral meetings and written submissions in response to a2005 Discussion Paper, a 2006 Discussion Guide and a 2008 Options for Possible Changepaper. Issue-specific working groups were established as follows: Working Group on Therapeutic Improvement; Working Group on International Therapeutic Class Comparison; Consultant and Working Group on Costs of Making and Marketing; Working Group on Price Tests; and Working Group on Patented Generic Medicines. The following summarizes the major issues under review, stakeholders´ views and the Board´s decisions on each of these issues.
1. Issue – Underlying Principles
In 2006-07, one of the elements of the consultations with stakeholders was whether the Board should be more explicit about the “Principles” that have guided its interpretation of the Patent Act (Act) as reflected in the Board´s Guidelines. Specifically, during the consultations a number of statements that various stakeholders have used in describing the Board´s role were listed and stakeholders were asked which, if any, best described their views on the mandate of the Board. They were also asked whether the Board should incorporate a statement of “Guiding Principles” into its Compendium of Policies, Guidelines and Procedures (Compendium). The principles were, in no particular order:
- Lowest Reasonable Price
- Canada should pay its Fair Share
- Value-based Pricing
- Simplicity/Transparency
- International Parity/Consistency
- Accessibility combined with Affordability
- Consistency over time – same test of “Excessiveness” applied over life of patent
Stakeholders' Views
At the five multi-lateral stakeholder meetings in the fall 2006, these principles were discussed. Views were varied as to which were most important but, in general, different principles were seen as important by different stakeholders.
Board's Position
In its May 31, 2007 Communiqué, the Board indicated that it was cognizant that the Government´s objective in creating the PMPRB was to ensure the additional patent protection provided to pharmaceutical patentees stemming from changes in the Act in 1987 did not translate into excessive prices. In keeping with this objective, the Board´s mandate is to ensure that prices charged by patentees for patented medicines sold in Canada are not excessive, thus protecting the interests of consumers. The Board supports the inclusion of language to this effect in the Compendium. With the change in the Table of Contents of the Compendium and the inclusion of a new section on Legal Framework, the reference to the Board´s consumer protection focus is discussed in that section rather than in the Preamble to the Guidelines.
2. Issue – Levels of Therapeutic Improvement
In response to the 2005 Discussion Paper on Drug Price Increases, stakeholders identified more pressing concern with regard to introductory drug prices. A key element of this concern was that the current approach of the Board in categorizing therapeutically comparable drug products does not recognize the current nature of incremental pharmaceutical innovation.
Stakeholders' Views
Stakeholders expressed mixed views. On the one hand, industry members thought that the categories were not needed and could be replaced by a clear definition of an “excessive price.” Other stakeholders supported the concept of assessing therapeutic improvement but thought changes to the current categories were warranted, particularly in regard to Category 3 – “moderate, little or no improvement” drugs.
A multi-stakeholder Working Group on Therapeutic Improvement was established to develop definitions or parameters relating to levels of therapeutic improvement and to provide advice on requirements for supporting evidence, and possible sources of such evidence.
Report of the Working Group on Therapeutic Improvement
In April 2008, the Working Group tabled its final report. It recommended a new series of four levels of therapeutic improvement and provided definitions for each level: Breakthrough; Substantial Improvement; Moderate Improvement; and Slight or No Improvement.
It also identified factors that would be considered to determine level of therapeutic improvement with the main factors being increased efficacy or reduction in the incidence or grade of adverse reactions, or savings to the Canadian health care system and/or to patients and/or caregivers. The Working Group proposed that the main factor to be used is therapeutic effect, which includes increased efficacy or reduction of side effects OR economic factors. Such economic factors include cost savings to the Canadian health care system (private or public payers and employers) and may or may not include cost savings to patients and/or caregivers. The Working Group proposed other factors to be considered in the evaluation of therapeutic improvement. Clinical factors include: duration of usual treatment course, success rate, geographic site of administration (e.g., institution (acute vs outpatient) vs home), percentage of affected population treated effectively, time required to achieve the optimal therapeutic effect, and route of administration (e.g., oral vs injectable). Economic/pharmaco-economic factors include: patient convenience/preference, caregiver convenience/preference, disability cost avoidance/savings, compliance improvements, and reduction in acute care or institutional health care costs.
The Working Group also proposed that certain factors not be considered in evaluating level of improvement: mechanism of action, a new chemical entity and a different pharmacokinetic profile.
Finally, it proposed using the Oxford Centre for Evidence-based Medicine Levels of Evidence as the basis for the scientific evaluation of new drug products.
Board's Position
The Board concurs with most of the recommendations of the Working Group, including four levels of therapeutic improvement and using the Oxford Centre´s levels of evidence. The Board also supports increased efficacy or improved safety (reduction in adverse reactions) as the primary factors to be considered. It agrees that secondary factors may be relevant and agrees with those proposed by the Working Group with the exception of the reduction in acute care or institutional health care costs, which it regards as difficult to clearly attribute as the impact of a particular new medicine. As well, it agrees that patient and caregiver convenience could be important, but not simply patient/caregiver preference. Finally, it supports the consideration of compliance improvements but only if this leads to improved therapeutic efficacy.
- Lowest Reasonable Price
- Canada should pay its Fair Share
- Value-based Pricing
- Simplicity/Transparency
- International Parity/Consistency
- Accessibility combined with Affordability
- Consistency over time – same test of “Excessiveness” applied over life of patent
Stakeholders' Views
At the five multi-lateral stakeholder meetings in the fall 2006, these principles were discussed. Views were varied as to which were most important but, in general, different principles were seen as important by different stakeholders.
Board's Position
In its May 31, 2007 Communiqué, the Board indicated that it was cognizant that the Government´s objective in creating the PMPRB was to ensure the additional patent protection provided to pharmaceutical patentees stemming from changes in the Act in 1987 did not translate into excessive prices. In keeping with this objective, the Board´s mandate is to ensure that prices charged by patentees for patented medicines sold in Canada are not excessive, thus protecting the interests of consumers. The Board supports the inclusion of language to this effect in the Compendium. With the change in the Table of Contents of the Compendium and the inclusion of a new section on Legal Framework, the reference to the Board´s consumer protection focus is discussed in that section rather than in the Preamble to the Guidelines.
3. Issue – International Therapeutic Class Comparison Test
The Board noted that the current Guidelines do not require Staff to review the prices at which other medicines in the therapeutic class are sold in countries other than Canada as per paragraph 85(1)(c) of the Act. However, this is a factor that the Board must consider should it need to determine if a price is excessive. As a first step, in its May 31, 2007 Communiqué, the Board announced that it would establish a small group of experts and interested stakeholders to develop a methodology for identifying appropriate therapeutically comparable medicines in comparator countries. It specified that the focus of the mandate for this group of experts would be based on scientific and clinical considerations only and would not include work on possible price tests nor on when or how this factor may be incorporated into price tests. The Working Group on Price Tests was subsequently mandated to provide advice on all price tests.
Working Group on International Therapeutic Class Comparison (ITCC)
The Working Group completed its report in April 2008. Its main recommendation was that the international therapeutic class be limited to those medicines identified in the domestic therapeutic class comparison.
Although its mandate was not to consider what price test might be associated with these comparator medicines sold internationally, and when such a test might be used in the price review, the Working Group did offer advice in this regard. It noted the difficulties in understanding the basis on which prices of other medicines are set in other countries and thus the reliability and utility of a test incorporating such prices would be poor. As a result, it recommended that this test should not normally be used, but could be conducted to provide additional information that may aid in the resolution of a dispute as to whether a price is excessive.
Working Group on Price Tests
The Working Group supported the recommendations of the ITCC Working Group, and further clarified two possible price test methodologies – the “Ratio Approach” and the “Straight Class Approach.”
Board's Position
The Board agrees that the result of an ITCC price test may not be reliable and that prices of medicines sold in other countries are the most remote in terms of the interests of Canadian consumers. However, this is a mandatory factor, under ss.85(1) of the Act, that the Board must consider in determining whether a price is excessive in the context of a hearing. The Board, therefore, considers it appropriate to add a Guideline on the conduct of the ITCC and associated price. Beyond hearings, this may also be of possible use to Board Staff and patentees in terms of another source of information in the context of an investigation.
4. Issue – Introductory Price Tests
In response to the 2005 Discussion Paper on Drug Price Increases, stakeholders responded that they were more concerned with high introductory drug prices, and saw these as a cost-driver.
Stakeholders' Views
Stakeholders recognized that introductory price tests are essentially currently tied to levels of therapeutic improvement. In general, industry believes that the Board should set one clear definition of what is an excessive price based on the price factors in ss. 85(1) of the Act. Others agreed that price tests should be associated with an assessment of the relative degree of therapeutic improvement.
In its May 31, 2007 Communiqué, the Board indicated that it was reserving comment on price tests in general, and their use, given it had decided to establish the Working Group on Therapeutic Improvement to provide advice on definitions of therapeutic improvement, and the ITCC Working Group to develop a methodology for identifying appropriate therapeutically comparable medicines in comparator countries.
Following the completion of the reports of these working groups, the Board established a Working Group on Price Tests.
Working Group on Price Tests
The Working Group recommended that increasing potential for price premium flexibility be associated with increasing levels of therapeutic improvement. They proposed a series of introductory price tests consistent with this principle for the four levels of therapeutic improvement: Breakthrough: Median of the International Prices (MIP); Substantial Improvement: Higher of the top of the Therapeutic Class Comparison (TCC) test and the MIP; Moderate Improvement: Higher of the mid-point between the top of the TCC test and the MIP, and the top of the TCC test; and Slight/No Improvement: Top of the TCC test.
The Working Group also agreed that, unless the patentee makes a submission on therapeutic improvement, line extensions will normally not be referred to HDAP and will be subject to the existing Reasonable Relationship (RR) test, and combination drug products will have a maximum non-excessive (MNE) price limited to the sum of the component ingredients if these are sold in Canada.
Board's Position
The Board agrees with the principle that opportunities for price premiums should be commensurate with the degree of therapeutic improvement and therefore the Board accepts the price tests proposed by the Working Group for the four new levels of therapeutic improvement. It also agreed to maintain the RR test for line extensions where no therapeutic improvement is proposed, and that prices of combination products would be limited to the sum of component products unless deemed to offer some additional therapeutic advantage.
5. Issue – Modified Guidelines for Certain Patented Generic Drug Products
Over the course of the Guidelines review, the Board met bilaterally with various stakeholder groups, including representatives of the generic industry. This sector of the pharmaceutical industry identified a number of issues that they argued are unique to the market dynamics and competitive forces faced by generics. They also argued that their prices are determined by public drug plans in reference to brand drug products with the same active ingredient. As well, such unique issues carry over into the comparator countries. As a result, they proposed that, for eligible generic drug products, certain specific departures from the normal Guidelines are reasonable.
Canadian Generic Pharmaceutical Association (CGPA)/PMPRB Working Group
The Working Group proposed certain modified Guidelines for two types of patented generic drug products: 1) those that are licensed versions of a patented brand medicine; and 2) those that are deemed by Health Canada to be bioequivalent to a reference brand drug product, even though it has a different processing/formulation patent. A third type of patented generic drug product, those with delivery or other types of patents that make them truly a new drug product, would be subject to the Guidelines applicable to all other patented drug products.
For the first two types of patented generics, the Working Group proposed that these not be referred to the Human Drug Advisory Panel (HDAP). Instead, the comparator drug products would automatically be determined as follows: generic drug products that are bioequivalent to a brand drug product would have as its sole comparator the reference brand; and those generic drugs that are a licensed version of an identical brand drug product (same patent, medicine, dosage form and strength) would have as its sole comparator that brand drug.
The Working Group also identified challenges in relation to international prices. In the case of a licensed patented generic drug product, the appropriate international prices would be those of the identical brand drug product. These prices may not be readily available to the generic company. Since the brand drug´s price in Canada must not be the highest in regard to the comparator countries, it follows that if the domestic price of the licensed generic is lower than the domestic price of the brand, it too will be lower than the international prices for that drug product.
For patented drugs that are bioequivalent, but not identical (due to a different processing patent) to a reference brand drug product, the appropriate international prices would be those of the patented generic drug product if it is sold in other countries, and not the prices of the reference brand. It is generally known, however, that prices of generic drugs tend to be higher than in comparator countries. The generic industry´s view is that the international generic market is very different from that in Canada and thus less relevant in terms of an appropriate price test. They proposed that the primary price test for a bioequivalent patented generic should be the TCC test using the reference brand drug product sold in Canada, regardless of whether the patented generic drug product´s price in Canada was the highest of the comparator countries.
Board's Position
The Board considers a streamlined TCC test, as proposed by the Working Group, in regard to bioequivalent and licensed patented generic drug products is reasonable. It also notes that such an approach would be less demanding of Board Staff time and not require the resources of HDAP.
In terms of international prices, all patentees are required to file international prices regardless of whether the company is regarded as innovative or generic. While this may be more difficult for licensed patented generic drug products because such information is not held by their own company, an effort in this regard is expected to be made particularly if the brand product is no longer sold in Canada and therefore not the primary comparator in the TCC test. However, the Board recognizes that, in general, giving primary weight to the TCC test is reasonable, given that if the brand drug product in Canada is not sold at an excessive price, by default it is not the highest among the comparator countries.
In the case of patented generic drug products that are bioequivalent to a brand drug product, it is possible, if not likely, that the Canadian price of the generic could be higher than the prices in comparator countries in which the same patented generic drug product is sold. While the Board has a “golden rule” that the price of the Canadian patented drug cannot be the highest in the world, in this particular case only the Board is prepared to give primary weight to the domestic TCC introductory price test, given that generally Canadian consumers already benefit from the fact that patented generic bioequivalent drugs fall under public drug plan pricing rules that make them significantly cheaper than the reference brand drugs. However, the “golden rule” is not waived if the reference brand drug product is not sold in Canada at the time the patented generic drug product is introduced. The above would also apply to price reviews in periods following the introductory benchmark period.
6. Issue – Impact of Reporting Benefits (De-linking of the ATP from the MNE Price)
In March 2007, the Federal Court (FC) issued a decision on a Judicial Review application in the matter of LEO Pharma Inc. and the medicine Dovobet, which included a ruling that the Patented Medicines Regulations (Regulations) made it explicit that the calculation of the Average Price or Net Revenue “shall” be net of all benefits, and that there was no discretion on the Board to differentiate as to whether free goods were of a compassionate nature. Significant concern was expressed by industry, which had been exercising considerable discretion to not report benefits they wished to have excluded, that forcing all benefits to be included in the Average Transaction Price (ATP) calculation would cause a significant price decrease with various negative consequences for the patentee. The industry further suggested that the Regulations be amended and/or that options be developed as part of the Guidelines review to mitigate the negative impacts and thus not create a disincentive for patentees such that they would no longer be prepared to offer benefits to customers.
In its January 2008 Discussion Paper, the Board asked for comments on potential options for amendments to the Regulations and related to a possible “de-linking” of the ATP from the CPI-Adjustment Methodology for the purposes of determining a Maximum Non-Excessive (MNE) price. One option dealt with the case of a declining ATP due to the offering of a benefit, while the second included the circumstance of an introductory ATP that was below the introductory MNE price.
Stakeholders' Views
In general, those that commented on the de-linking concept supported the idea of enabling an actual price to go up to, or “return” to, a previous price that had been determined to not be excessive. However, non-industry respondents argued that their support for the option was contingent on some constraint being placed on the maximum amount a price could increase in any single year.
Working Group on Price Tests
The Working Group reviewed the current CPI-Adjustment Methodology, and proposed two exceptional circumstances where the ATP would be “de-linked” from the normal CPI methodology, which represent a slight modification of the options discussed in the January 2008 Discussion paper. These are referred to as the “GAP” situation (GAP) and the “DIP” situation (DIP).
A GAP exists when the introductory ATP of the medicine is below the MNE price or the patentee reduces its price below a previously non-excessive ATP in response to market forces. The GAP, therefore, means the difference between the current ATP and the introductory MNE price or the current ATP and a previous non-excessive ATP where the drop in price is not due to a benefit. Since the amount of the GAP could be significant, the Working Group also agreed to a threshold limit on the amount of any single year price increase – 33% of the price gap, not to exceed either 10% or 15% of the actual year´s ATP, which the threshold to be determined by the Board.
A DIP refers to a situation where a patentee offers a new or enhanced benefit to a customer which causes its net ATP to decline from a previous non-excessive price. The Working Group proposed that, when the benefit is ended and the patentee submits evidence to Board Staff that the subsequent increase in price was solely due to the end of a benefit, the new ATP would not be considered excessive as long as it did not exceed, or “rebound” higher than, the previous non-excessive price.
Beyond these two exceptions, the Working Group did not come to agreement on any other changes to the current CPI-Adjustment Methodology but did consider two options:
- Retain the 3 year “banking” but to eliminate the one year “cap”; or
- Eliminate banking and allow only simple CPI each year.
Board's Position
The Board does not wish to unduly create a disincentive to the offering of benefits to customers. In regard to the exceptions proposed by the Working Group, the Board considers it reasonable that, in a circumstance where an apparent excessive price is due solely to the termination of a benefit and the patentee provides evidence in this regard (i.e., the DIP situation), the resulting ATP should not be viewed as excessive if it simply “rebounds” to the pre-benefit price, which is the price that other customers that did not receive the benefit would have continued to pay.
The Board is concerned about the potential price increases that could result from the proposed CAP methodology. This is a matter that the Board feels requires further analysis, particularly once the calculation of the ATP net of all benefits associated with a sales transaction become mandatory for the January to June 2009 reporting period and beyond.
As part of its Notice and Comment, the Board is interested in hearing views on the “cap” factor.
7. Issue – Any Market Price Reviews
Part of the concern of stakeholders with respect to introductory prices was that the ability of some classes of customers or provincial jurisdictions to obtain favourable prices might result in other customers/jurisdictions paying a price that would be excessive.
Stakeholders' Views
In 2007, stakeholders generally supported the use of the average national price both on the ground of efficiency and the high degree of compliance with the Guidelines. However, others were concerned about price equity and that the Board´s role was to ensure that no relevant market paid an excessive price. In general, all agreed that reviews at the level of “any market” should be done on a case-by-case basis, where warranted.
In the January 2008 Discussion Paper, the Board sought feedback on four circumstances when a price review at the level of any market would be conducted. One of these circumstances was at the time of the review of the introductory price. The second situation was as part of an investigation into an excessive price, as appropriate – see Chapter 3 Investigations. As well, it would be done in response to a complaint. Industry was generally opposed to the circumstances, and viewed the approach as unwarranted and unnecessary. On the other hand, consumers, governments and others thought that the PMPRB should exercise its authority to undertake price review in any market in order to limit significant price disparities.
Working Group on Price Tests
The Working Group on Price Tests agreed that any market price reviews should only be conducted at introduction so that a price for any class of customer (i.e., pharmacy, hospital and wholesaler) would not exceed the national introductory maximum non-excessive price.
For existing drug products, the PMPRB should only undertake any market price reviews in case of an investigation where variability in average prices in different markets (class of customer or province/territory) appears to be an issue.
Should a price appear excessive in any market, the Working Group discussed two options regarding the appropriate approach in calculating the excessive revenues but did not reach an agreement.
- Excess revenue would be based on the average price across all markets in Canada (national ATP) and not just on the excess revenue for the market where the price is excessive. This approach takes into account foregone revenues in markets where benefits were offered.
- Excess revenue would be calculated based only on the market where the price was excessive.
Board's Position
The Board´s mandate is to ensure that prices of patented medicines sold in Canada are not excessive in any market in Canada. The Board believes that it is important to ensure that introductory prices are not excessive for any class of customer or in any province/territory. For existing drug products, the Board agrees that any market price reviews only be carried out on a case-by-case basis where price variability in different markets appears to be an issue.
As part of this Notice and Comment, the Board is interested in hearing views on the appropriate methodology for calculating excess revenues arising from a price that is excessive in a market in Canada.
8. Issue – Re-setting the MNE Price
In 2006, stakeholders were consulted on whether the Board should effectively “re-set” the maximum non-excessive price (MNE) rather than continue to only allow price increases related to the Consumer Price Index. In the January 2008 Discussion Paper, three circumstances were proposed when “re-setting” the MNE price might be appropriate: 1) in the case of new scientific information; 2) when the new drug product was sold in too few countries and the Median International Price was the pivotal introductory price test; and 3) potential cost of making and marketing arguments.
Stakeholders' Views
In general, industry did not support the proposed circumstances on the basis that the proposals would limit the circumstances in which a price could be re-set and also increase price uncertainty. Others were more supportive of these provisions, but all stakeholders wanted more clear definitions for the costs of “making” and “marketing”, and the “triggers”, particularly in terms of new scientific information.
Working Group on Price Tests
The Working Group agreed that in the event a price is re-set downward, e.g., because Canada has become the highest-priced country among the comparator countries, a patentee should have a full year, or to the end of the following calendar year, to reduce its price to the new MNE price, without the commencement of an investigation or the calculation of excess revenues.
Expert and Working Group on ss. 85(2) – “Costs of Making and Marketing”
An expert economist was contracted to prepare a report analyzing issues related to the definitions of “costs of making and marketing”. The main conclusions of the economist, that were generally supported by the Working Group, were that cost-based price regulation is difficult for many reasons (including uncertainty about what costs to include, difficulties in verifying cost information that is generally not in the public domain, etc.), and should be avoided where possible.
If some cost-based price regulation became necessary, it was recommended that it be considered in cases of price changes rather than initial prices. It was also proposed that two particular circumstances might be more amenable to assessing costs: 1) where there was direct evidence of clinically-relevant drug improvements which were accompanied by additional costs; and 2) exceptional cost increases resulting from unanticipated and non-transitory events (e.g., new post-market requirements imposed by a regulator, sustained shortages in the supply of the ingredient medicine), which were beyond the control of the company.
Finally, it was recommended by the expert that “jurisprudence” may be preferable to guidelines given the difficulties involved, and the likely unique aspects of each case.
Board's Position
In terms of the specific circumstances for re-setting the MNE price, the Board agrees that the current approach to reviewing the Median International Price (MIP) within three years or once the drug product is sold in five comparator countries is appropriate.
In terms of scientific issues, the Board supports the identification by the HDAP of any important weaknesses or gaps in scientific evidence. However, the Board does not believe that a specific Guideline is needed, and prefers to leave the discretion to Board Staff in the application of the review process on a case-by-case basis, as appropriate.
If such downward price setting occurs, the Board accepts the recommendation that the patentee be given to the end of the following calendar year to ensure that the price does not exceed the new MNE price.
The Board notes that reviewing the costs of making and marketing may be considered, but is not mandatory, and would only be considered if the Board is unable to determine if the price of a patented drug product was excessive based on the factors in ss. 85(1). Therefore, it has decided not to propose a specific Guideline in this regard.
Summary of Proposed Changes
In light of the Board´s decisions, the draft revised Guidelines and Procedures include additions and revisions in 6 key areas:
1. Four new levels of therapeutic improvement which replace the previous three categories.
2. Modified price tests aligned with the four levels of improvement;
3. Alternate processes for bioequivalent and licensed patented generic drug products regarding the selection of comparable drugs and the conduct of the TCC Test and the IPC Test.
4. Guidelines on the conduct of selected any market price reviews at introduction and, thereafter, on a case-by-case basis, as appropriate.
5. Guidance on the possible conduct of an International Therapeutic Class Comparison Test.
6. A new exceptional De-Linking Methodology (DIP situation) to be used.
In addition, the Board is seeking comments on : – CPI cap factor; and – The methodology for calculating excess revenue arising from excessive price in any market.
The Guidelines and Procedures section has also been reorganized into an order that follows the actual stages of the review process (i.e., the scientific review process, the price review process, and the investigations process). The schedules are also reordered to mirror the sequence in which they are used.
In addition, the Compendium itself has been expanded to include a new section on the Legal Framework within which the PMPRB operates, as well as a section summarizing the major Policies that the Board has adopted.
Preamble
The Patented Medicine Prices Review Board (PMPRB) is committed to making the price review process more open and transparent to all stakeholders.
One of the primary objectives of the Compendium of Policies, Guidelines and Procedures (Compendium) is to ensure that patentees are aware of the policies, guidelines and procedures under which Board Staff reviews the prices of patented medicines sold in Canada, and the procedures normally undertaken in the scientific and price review and when a price appears to be excessive.
From time to time, the PMPRB finds it necessary to update the Guidelines under which it operates to ensure that they remain relevant and appropriate. When considering Guidelines amendments, the PMPRB consults with its stakeholders through its Notice and Comment process. This draft version of the Compendium was published on August 20, 2008.
The Compendium is organized as follows:
I - Legal Framework
II - Policies
III - Guidelines and Procedures
Chapter 1 - The Scientific Review Process
Chapter 2 - The Price Review Process
Chapter 3 - Investigations
Chapter 4 - Schedules
I - Legal Framework
1. Origin of the PMPRB
1.1 The PMPRB was established pursuant to amendments to the Patent Act (the Act) that came into force on December 7, 1987. Prior to 1987, Canada sought to moderate the prices of patented medicines by means of compulsory licenses to increase competition. Under the 1987 amendments, Canada strengthened patent protection of medicines to provide patentees with an incentive to invest in more pharmaceutical research and development (R&D) in Canada. These amendments also established the PMPRB as the consumer protection pillar of the pharmaceutical patent law reform to ensure that the prices of patented medicines are not excessive.
1.2 Further amendments to the Act, which came into force on February 15, 1993, abolished the granting of compulsory licenses for patented medicines to generics so that patentees have market exclusivity for the entire term of their patents. In order to fill the vacuum created by the abolition of compulsory licenses, these amendments also expanded the PMPRB's remedial powers so that it could now order payment of any excess revenues derived by patentees while selling a medicine at an excessive price, in addition to ordering price reductions. Patentees may also be subject to fines or imprisonment for failure to comply with the PMPRB´s orders.
2. Mandate of the PMPRB
2.1 The PMPRB has the following dual mandate:
- Regulatory - To ensure that the prices charged by patentees for patented medicines sold in Canada are not excessive, pursuant to sections 83 and 85 of the Act, thereby protecting consumers and contributing to Canadian health care; and
- Reporting - To report annually to Parliament on its activities, on the ratios of R&D expenditures to sales by the patented pharmaceutical industry and by individual patentees, and on pricing trends within the pharmaceutical industry, relating to all medicines, pursuant to sections 89 and 100 of the Act, thereby contributing to informed decisions and policy making in health care; other reporting activities include inquiring into and reporting on any matter that the Minister of Health refers to it, pursuant to section 90 of the Act.
3. Structure and Operation of the PMPRB
3.1 The PMPRB is an independent and autonomous quasi-judicial body. To ensure this independence and autonomy, the Act provides no power, either expressly or implicitly, to the government to direct the PMPRB or to review its decisions and orders. The PMPRB also has no involvement in federal policy-making. However, decisions of the PMPRB are subject to judicial review by the Federal Court on jurisdictional or procedural grounds in accordance with administrative law principles.
3.2 The PMPRB is composed of Board members, appointed pursuant to subsection 91(1) of the Act, and staff (Board Staff), appointed pursuant to subsection 94(1) of the Act.
3.3 The PMPRB has the authority to develop policies and procedures as to how it will carry out its statutory duties in a fair and effective manner. Part of the process by which the PMPRB has determined to carry out its statutory obligations is by the administrative separation of its review and prosecutorial functions, performed by Board Staff on the one hand, and its adjudicative function performed by Board members on the other hand.
3.4 Board Staff carries out the day-to-day work of the PMPRB including the administration of the Patented Medicines Regulations (the Regulations) to ensure compliance with the prescribed filing requirements and the review of prices of patented medicines in accordance with Guidelines of the Board.
3.5 If the Chairperson of the Board decides that it is in the public interest that a hearing be held, pursuant to subsection 83(6) of the Act, to determine whether a patented medicine is being or has been sold at an excessive price in any market in Canada, the Chairperson will issue a Notice of Hearing and will appoint a panel of Board members to preside at the hearing (Hearing Panel).
3.6 To preserve the impartiality of Board members, until a matter is brought before a Hearing Panel at a public hearing, no Board member is informed of the results of Board Staff´s review into an instance of possible excessive pricing, other than the Chairperson in his management capacity as the Chief Executive Officer of the PMPRB, pursuant to subsection 93(2) of the Act, which is done solely for the purpose of determining whether a hearing is in the public interest.
4. Jurisdiction of the PMPRB Pertaining to Price Regulation
4.1 Sections 79 and 85 of the Act give the PMPRB jurisdiction to determine whether a patentee of an invention pertaining to a medicine is selling or has sold the medicine at an excessive price in any market in Canada if the following criteria are satisfied:1
4.1.1 Patentee or Former Patentee
- A party must be a patentee of an invention. Subsection 79(1) of the Act defines a “patentee” as a person for the time being entitled to the benefit of a patent for an invention, including any other person entitled to exercise rights in relation to the patent, with the exception of a person granted a compulsory license by the Commissioner of Patents before December 20, 1991 that was not terminated before the day amendments to the Act came into force on February 15, 1993.
- The PMPRB also has jurisdiction over a former patentee of an invention, while it was a patentee, but its jurisdiction does not extend to a former patentee who has not been entitled to the benefit of the patent or to exercise any rights in relation to the patent for three or more years.
4.1.2 Patent pertains to the medicine
Medicine
- A medicine is defined as any substance or mixture of substances made by any means – whether produced biologically, chemically or otherwise – that is applied or administered in vivo in humans or in animals to aid in the diagnosis, treatment, mitigation or prevention of disease, symptoms, disorders, abnormal physical states, or in modifying organic functions in humans or animals, however administered.
- For greater certainty, this definition includes vaccines, topical preparations, anaesthetics and diagnostic products used in vivo, regardless of delivery mechanism (e.g., transdermally, capsule form, injectable, inhaler, etc.). This definition excludes medical devices, in vitro diagnostic products and disinfectants that are not used in vivo.
Patent
- Subsection 79(2) of the Act provides that a patent for an invention pertains to a medicine if the invention is intended or capable of being used for medicine or for the preparation or production of medicine.
- On the face of the patent, there must be a rational connection or nexus between the invention described in the patent and the medicine, which can be one of the merest slender thread.
4.1.3 Sale in any market in Canada
- Pursuant to section 85 of the Act, the patentee or former patentee must be selling or have sold the patented medicine in any market in Canada.
- With the exception of medicines sold under compulsory licenses granted by the Commissioner of Patents before December 20, 1991 that were not terminated before the day amendments to the Act came into force on February 15, 1993, all patented medicines sold in any market in Canada for human or veterinary use are covered by the PMPRB's price review jurisdiction, including patented medicines sold pursuant to Notices of Compliance, under the Special Access Programme, through Clinical Trial Applications, or as Investigational New Drugs.
- The PMPRB reviews the ex-factory prices established for the first sale of a patented medicine at arm´s-length by the patentee, directly to a class of customer, namely a wholesaler, hospital, pharmacy or other. The PMPRB has no authority over prices charged by wholesalers or retailers or over pharmacists´ professional fees.
- Prices do not need to be approved by the PMPRB before patented medicines are sold in Canada. The PMPRB does not set the prices at which patented medicines can be sold but determines the maximum non-excessive (MNE) prices at which these medicines can be sold in Canada.
5. Price Regulation Factors
5.1 Subsection 85(1) of the Act stipulates those factors that the Board, during the course of a hearing, must take into consideration when determining whether a patented medicine is being sold or has been sold at an excessive price in any market in Canada by a patentee or former patentee. These factors are:
- the prices at which the medicine has been sold in the relevant market;
- the prices at which other medicines in the same therapeutic class have been sold in the relevant market;
- the prices at which the medicine and other medicines in the same therapeutic class have been sold in countries other than Canada;
- changes in the Consumer Price Index; and
- such other factors as may be specified in any regulations made for the purposes of this subsection.
5.2 If after considering the above factors, the Board is unable to determine if a price is excessive, subsection 85(2) of the Act stipulates that it may consider the costs of making and marketing the medicine as well as other factors which can be specified by regulations or that the Board considers relevant in the circumstances.
5.3 The Board, following considerable deliberation and consultation with all stakeholders, pursuant to subsection 96(5) of the Act, published the PMPRB´s Guidelines pursuant to subsection 96(4) of the Act. Although the Guidelines are not binding on the Board or the patentee, they establish an approach and methodology in applying the factors set out in subsection 85(1) of the Act.
6. Filing Requirements Pertaining to Price Regulation
6.1 The PMPRB must have timely and accurate information to fulfill its regulatory mandate.
6.2 The Act and the Regulations set out the filing requirements pertaining to price regulation for a patentee or former patentee of an invention pertaining to a medicine that falls under the jurisdiction of the PMPRB. Further details on each element of information to be reported, and how and when the information is to be submitted to the PMPRB, can be found in the Patentee´s Guide to Reporting Forms 1, 2 and 3 pursuant to the Patented Medicines Regulations.
Notification of Intention to Sell a Patented Medicine
- Section 82 of the Act requires a patentee to notify the PMPRB of its intention to offer a patented medicine for sale in a market in Canada in which it has not previously been sold, and of the date on which sales are expected to begin, as soon as it is practicable to do so.
- The Board may order a patentee to provide information relating to the price at which it intends to sell the patented medicine. However, information relating to the price need not be provided earlier than 60 days before the date on which the product is intended to be sold.
Form 1 (Medicine Identification Sheet)
- Subsection 3(1) of the Regulations requires a patentee or former patentee of an invention pertaining to a medicine to report to the PMPRB prescribed information identifying the patented medicine (Form 1). Form 1 is required for all patented medicines for human or veterinary use and shall be accompanied by the product monograph for the patented medicine or, if an NOC has not been issued in respect of the patented medicine, by information similar to that contained in a product monograph.
- Subsections 3(2) and 3(3) of the Regulations require that Form 1 information must be reported if an NOC has been issued in respect of the medicine or the medicine is being offered for sale in Canada, within seven days after the day on which the first NOC is issued in respect of the medicine, or within seven days after the day on which the medicine is first offered for sale in Canada, whichever comes first.
- If a patentee or former patentee begins selling a medicine in Canada during the pre-grant infringement period, once the patent is issued, the patent or former patentee is required to file Form 1 information with the PMPRB.
Form 2 (Information on the Identity and Prices of the Medicine)
- Subsection 4(1) of the Regulations requires a patentee or former patentee of an invention pertaining to a medicine, which is selling or has sold the medicine in any market in Canada, to report to the PMPRB prescribed information identifying the medicine and concerning the price of the medicine (Form 2). This includes the date on which the medicine is first sold in Canada and the quantity of medicine sold in final dosage form, and either the average price per package or net revenues from the sales of each dosage form, strength and package size in which the medicine was sold by the patentee or former patentee to each class of customer in each province and territory.
- Subsection 4(4) of the Regulations provides that, in calculating the average price per package or net revenues, the actual price or actual revenue after any reduction including rebates, discounts, refunds, free goods, free services, gifts or any other benefit of a like nature shall be used.
- Subsection 4(2) of the Regulations requires that, if the medicine is for human use and contains a controlled substance as defined in the Controlled Drugs and Substances Act, such as opioids, amphetamines, barbiturates and benzodiazepines, or is a substance listed or described in Schedules C or D of the Food and Drugs Act, such as radiopharmaceuticals, vaccines, blood products and insulins, or is listed or described in Schedule F of the Food and Drug Regulations, such as medicines requiring a prescription, the prescribed information under Form 2 must be reported within 30 days after the day on which the medicine is first sold in Canada (for the first day´s sales), and within 30 days after each six month period commencing on January 1 and July 1 of each year, in respect of each of these periods, including the final partial period during which the reporting party exercises rights under the patent.
- Subsection 4(3) of the Regulations requires that, for medicines for human use that do not contain a controlled substance or do not contain a substance listed or described in the schedules listed in subsection 4(2), including non-prescription medicines for human use or for all medicines for veterinary use, the prescribed information under Form 2 must be reported within 30 days after the date on which the PMPRB sends a request in response to a complaint for each six month period commencing on January 1 and July 1 of each year, and during the two years following the request, within 30 days after each 6 month period. A patentee or former patentee shall maintain up to date Form 2 information from the date of first sale in the event of a request for this information from the PMPRB in response to a complaint.
- A patentee or former patentee who does not voluntarily file Form 2 information on a medicine being sold during the pre-grant infringement period is required to ensure that this information is kept up-to-date for ultimate submission to the PMPRB, upon the issuance of the patent pertaining to the medicine.
6.3 All required information referenced in Paragraph 6.2, must be submitted using the appropriate electronic documents made available on the PMPRB´s Web site, under the heading "Legislation, Regulations and Guidelines - Patentee's Guide to Reporting". The completed electronic document, in its original format and file type, must be sent to the e-mail address specified on the PMPRB´s Web site.
6.4 The electronic documents submitted to the PMPRB must bear the electronic signature of an authorized individual, certifying that the information set out in the document is true and complete.
7. Consequences of Failure to File Required Information Pertaining to Price Regulation
7.1 Evidence of failure to file a Notification of Intent to Sell a Patented Medicine, pursuant to subsection 82(1) of the Act, may be brought to the attention of the Chairperson who may issue an order requiring production of this information.
7.2 If a patentee or former patentee, as the case may be, fails to file some or all of its Form 1 or Form 2 information for one or more periods by the regulatory deadlines, it will be advised in writing by Board Staff that it is in failure to file and be given seven days from the date the letter is sent out to file the missing information. If the patentee or former patentee does not comply, without further notice Board Staff will bring a motion before the Chairperson seeking a Board Order, pursuant to section 81 of the Act, requiring the patentee or former patentee to file the information within such time as is specified in the order.
7.3 If it appears to the Chairperson or to the Board that the patentee or former patentee failed to file information pursuant to section 80, or subsections 82(1) of the Act, or failed to comply with a Board Order made pursuant to section 81, or subsection 82(2) of the Act, to avoid or frustrate the mandate of the PMPRB, including to escape or delay the review of the price at which a patented medicine is sold in Canada, the Board may refer the matter to the Attorney-General of Canada to determine if summary conviction proceedings should be commenced under subsection 76.1(1) of the Act.
7.4 A Board Order made under section 81 or subsection 82(2) may be made an order of the Federal Court or any superior court of a province, enforceable in the same manner as an order of the court under section 99 of the Act.
8. Protection of Confidential Information Pertaining to Price Regulation
8.1 Pursuant to subsection 87(1) of the Act, apart from the exceptions noted below, any information or document provided to the PMPRB under section 80, 81 or 82 of the Act or in any proceeding under section 83 is privileged, and cannot be disclosed without the authorization of the person who provided it, unless it has been disclosed at a public hearing under section 83.
8.2 Any information that a patentee or former patentee submits to the PMPRB that is in the public domain will not be considered privileged under subsection 87(1) of the Act. This information may include publicly available product monographs, clinical trials or practical guidelines.
8.3 The privilege provided under subsection 87(1) does not extend to information and materials collected by the PMPRB, including any analysis performed by Board Staff of that information.
8.4 Subsection 86(1) of the Act gives the Board discretion to hold a hearing, or any part of it, in private if satisfied on representations from the patentee that specific, direct and substantial harm would be caused to the patentee by the disclosure of information or documents at a public hearing.
8.5 Information on the status of the price review by Board Staff, including the compliance status of patentees, is not protected by privilege under subsection 87(1) and may be made publicly available. As such, the PMPRB will publish summary reports on the results of the price review for all new active substances. The PMPRB may publish the results of other reviews.
8.6 Under subsection 87(2) of the Act, any information or document provided to the PMPRB under section 80, 81 or 82 of the Act, or in any proceeding under section 83, may be disclosed to any person engaged in the administration of the Act under the direction of the Board; to the Minister of Industry or other federal Minister designated by the Regulations; and to the provincial ministers of health and their officials, for the purpose of making representations to the Board with respect to a hearing under section 83.
II - Policies
Introduction
From time to time, the Board finds it necessary to adopt policies to indicate to stakeholders the principles it applies when interpreting its mandate. The following is a consolidation of the key policies that have been decided on by the Board. While the following policies are not binding on the Board, they help to promote consistency and transparency for stakeholders.
1. Patent Pertaining Policy
1.1 For the purposes of its jurisdiction, the PMPRB considers as a patent, any Canadian patent of invention that pertains to a medicine. This includes, but is not restricted or limited to, patents with the following status:
- patents for active ingredients;
- patents for processes of manufacture;
- patents for a particular delivery system or dosage form that are integral to the delivery of the medicine;
- patents for indications/uses; and
- patents capable of being used, whether or not they are being worked.
2. Patent Pending Policy
2.1 When a medicine subject to a pending patent is being sold in any market in Canada, the PMPRB will, when the patent is issued, review the price as of the date of first sale or the date on which the patent application was laid open, whichever comes later.2 Once the patent is granted, the PMPRB´s jurisdiction over the price at which the medicine was sold extends to the pre-grant infringement period, as the party selling the medicine derives the benefit of the patent during this period and so is a "patentee", pursuant to subsection 79(1) of the Act.
3. Patent Dedication Policy
3.1 The PMPRB will continue to assert jurisdiction over the price at which a patented medicine is sold in any market in Canada after the patent has been dedicated until the cancellation or surrender of the patent pursuant to the express provisions of the Act or the expiry of the term of the patent. The Act, which is the mechanism by which the state grants patents, and which confers rights and benefits for the duration of the term of the patent, does not expressly recognize patent dedication as a mechanism by which patent rights may be terminated before the normal expiry of the patent term.
4. Policy for when a Price Exceeds the Guidelines
4.1 The price of a drug product will be considered to have exceeded the Guidelines if its Average Transaction Price (ATP) exceeds its MNE price.
4.2 If the ATP exceeds the MNE price in a given period, but does not trigger the criteria for commencing an investigation, the patentee will be notified and the drug product will be reported as "Exceeds the Guidelines." The patentee will be expected to decrease its price and offset the excess revenues.
4.3 If the ATP is found to exceed the MNE price by an amount which triggers the investigation criteria, the patentee will be notified of the commencement of an investigation and the drug product will be reported as "Under Investigation." See Section III, Schedule 6.
5. Policy on Unit of Price Review
5.1 The PMPRB reviews the average price of each strength of an individual, final dosage form of each patented medicine sold in Canada, including:
- Drugs which have been assigned a Drug Identification Number (DIN) by Health Canada
- Drugs available under the Special Access Programme
- Drugs available through a Clinical Trial Application and
- Investigational New Drugs
5.2 Each strength of an individual, final dosage form of a patented medicine is referred to as a “drug product” throughout this Compendium.
5.3 The price of a drug product will normally be expressed as the price per unit in which that drug product is sold (i.e., tablet, millilitre, inhaler, etc.) rounded to the fourth decimal place.
III - Guidelines and Procedures
Preface to the Board´s Guidelines & Procedures
The following guidelines and procedures represent direction from the Board, to patentees and Board Staff, in order to provide assistance on how to comply with the Patent Act and the Patented Medicines Regulations. Please note: These Guidelines are not binding on patentees or on the Board in the context of a hearing.
The following section is organized as follows:
Chapter 1 - The Scientific Review Process: An evidence-based process that assesses the level of therapeutic improvement of a patented drug product and determines, where appropriate, the comparable drug products, dosage forms and dosage regimens, or drug products that are superior or inferior in terms of therapeutic improvement.
Chapter 2 - The Price Review Process: Once determined, the level of therapeutic improvement of a patented drug product, along with its comparators, are used in the appropriate PMPRB price test(s) to determine the Maximum Non-Excessive (MNE) price for the introductory period of the new drug product. Following introduction, the price of an existing medicine is reviewed according to the relevant price tests.
Chapter 3 - Investigations: The guidelines used and procedures undertaken when a price appears to be outside of the Guidelines.
Schedules: These Schedules are appended to the Guidelines in order to provide additional technical detail on a number of topics, and should be regarded as detailed procedures on the application of the Guidelines.
Chapter 1 - The Scientific Review Process
1. Introduction
1.1 The PMPRB's scientific review is an evidence-based process that recommends the level of therapeutic improvement of a patented drug product.
1.2 The scientific review process for all drug products (including those available through the Special Access Programme, Clinical Trial Applications and as Investigational New Drugs), will be undertaken following the guidelines and procedures in this chapter.
2. Sources of Scientific Information
2.1 The scientific review of a drug product is based on information provided by a variety of sources, including:
- Patentees may provide Board Staff with a brief submission (see Schedule 1) which clearly explains the rationale for the patentee´s proposals relative to level of improvement, comparable drug products and comparable dosage regimens.
- Research by a Drug Information Centre - Board Staff uses the services of a drug information centre to carry out research of scientific information such as clinical trial information, clinical practice guidelines, etc. The basis of the review by the drug information centre is the product monograph or information similar to that contained in a product monograph.
- Research by Board Staff - Board Staff may also update research and supplement data and evidence for a review.
- Members of the Human Drug Advisory Panel (HDAP) may also undertake their own research and supplement evidence for a review.
3. Human Drug Advisory Panels
3.1 The Human Drug Advisory Panel was created to provide expertise and advice to Board Staff in conducting the scientific review. HDAP performs the following functions:
- review and evaluate scientific information;
- consider advice from other experts (when deemed necessary);
- recommend, by majority decision, the level of therapeutic improvement of the new drug product, clinically equivalent medicines or, if none, other medicines that are superior or inferior in treating the same indications/use, and comparable dosage forms and dosage regimens where possible; and
- identify significant uncertainties in the evidence which may affect the analysis on which its recommendations are based.
3.2 In general, new drug products are referred to HDAP. However, the following drug products will not be referred to HDAP unless the patentee files a submission claiming therapeutic improvement:
- the new drug product represents a new DIN of an existing dosage form of an existing medicine, or a new DIN of another dosage form of the medicine that is comparable to the existing dosage form as per Schedule 3; or
- the new drug product is a combination drug product, the individual components of which are sold in Canada; or
- the new patented generic drug product is considered by Health Canada to be bioequivalent to the reference brand medicine sold in Canada; or
- the new patented generic drug product is a licensed version of an existing brand drug sold in Canada.
Procedures:
3.3 HDAP is composed of members with recognized expertise in drug therapy who have experience in clinical research methodology, statistical analysis and the evaluation of new drugs.
3.4 HDAP and its individual members do not meet with patentees.
3.5 The names of the members of HDAP are posted on the PMPRB's Web site.
3.6 The dates of HDAP meetings are posted on the PMPRB´s Web site.
3.7 At the request of a patentee, a drug product will also be referred to HDAP to provide pre-advisory assistance (pre-sale and/or pre-patent advisory assistance).
3.8 For drugs referred to HDAP, a patentee must file a submission which contains the elements referred in Schedule 1 at least two months prior to an HDAP meeting.
3.9 In the event that a large number of submissions are received for any one HDAP meeting, priority will be determined as follows:
- Drug products that are patented and sold;
- Drug products that are patented and about to be sold;
- Drug products that are patented but not sold;
- Drug products that are not patented but sold;
- Drug products that are not patented and are not sold.
3.10 The patentee will be advised of the date of the HDAP meeting at which its submission will be considered.
3.11 The HDAP report will include recommendations on the level of therapeutic improvement, comparator medicines (clinically equivalent or superior/inferior) and comparable dosage regimens, as well as an explanation of how the primary and secondary factors (see Paragraph 6) were applied and the evidence (see Paragraph 7) used.
3.12 A copy of the HDAP report will be sent to the patentee following the meeting of HDAP.
4. Determining the Primary Indication/Use of a Drug Product
4.1 Determining the primary approved or proposed indication, or primary indication/use if not approved for market in Canada, is important for the assessment of the level of therapeutic improvement of a new drug product with multiple approved indications/multiple uses. Procedures:
4.2 The level of therapeutic improvement for new drug products with multiple approved indications or multiple uses will be based on the approved indication or use for which the drug product offers the greatest therapeutic advantage in relation to alternative therapies for the same indication/use in a significant patient population. This would exclude rare medical conditions or diseases (i.e., low incidence and prevalence in Canada).
4.3 This approved indication or use will be considered the "primary indication/use" for the purposes of selecting comparable drug products.
4.4 Where there is no apparent single approved indication or use for which the drug product offers the greatest therapeutic advantage, the approved indication or use representing, potentially, the greatest proportion of sales will be the basis for determining its level of therapeutic improvement and selection of comparable drug products.
4.5 Estimates of potential sales can be based on several sources including actual prescribing patterns (when available), epidemiological data (Canadian incidence and prevalence) and prescribing patterns in other countries.
5. The Level of Therapeutic Improvement
5.1 HDAP utilizes the following set of definitions to determine the level of therapeutic improvement of a drug product:
Breakthrough: A breakthrough drug product is the first one to be sold in Canada that treats effectively a particular illness or clinical indication/use.
Substantial Improvement: A drug product offering substantial improvement is one that, relative to other drug products sold in Canada, provides substantial improvement in therapeutic effects.
Moderate Improvement: A drug product offering moderate improvement is one that, relative to other drug products sold in Canada, provides moderate improvement in therapeutic effects.
Slight or No Improvement: A drug product offering slight or no improvement is one that, relative to other drug products sold in Canada, provides slight or no improvement in therapeutic effects.
6. Factors Considered in Determining the Level of Therapeutic Improvement
6.1 The following factors are to be used in determining the level of therapeutic improvement of a drug product:
Primary Factors
- Increased efficacy
- Reduction in incidence or grade of important adverse reactions Secondary Factors
- Duration of usual treatment course
- Success rate
- Percentage of affected population treated effectively
- Time required to achieve the optimal therapeutic effect
- Route of administration
- Patient convenience
- Compliance improvements leading to improved therapeutic efficacy
- Caregivers convenience
- Disability avoidance/savings
6.2 The primary factors will be given the greatest weight, followed by an assessment of any additional improvement as a result of the secondary factors.
6.3 In determining the level of therapeutic improvement of new drug products, factors such as the following will generally not be taken into consideration, unless the impact of these factors results in either increased efficacy and/or a reduction in the incidence or grade of important adverse reactions:
- The mechanism of action
- A new chemical entity
- A different pharmacokinetic profile
Procedures:
6.4 The level of therapeutic improvement will be determined by the available evidence for the primary factors in order to assess if the drug product represents a breakthrough, substantial, moderate or little or no improvement relative to other drug products available in Canada.
6.5 Other factors will be considered on a case by case basis in addition to the primary factors. These factors will be weighed by HDAP based on sound evidence and reasonable clinical judgement.
6.6 These secondary factors could result in the level of therapeutic improvement being assessed at up to one level above that assessed by primary factors alone (i.e., from Slight or No Improvement to Moderate Improvement or from Moderate Improvement to Substantial Improvement).
7. Methodology for the Evaluation of the Level of Therapeutic Improvement
7.1 An evidence-based approach will be used to assess the drug under review using the hierarchy of evidence from the Oxford Centre for Evidence-Based Medicine (see Schedule 2).
Procedures:
7.2 HDAP will critically appraise the evidence obtained with regards to validity, impact and applicability. Level 1 evidence will be weighted higher compared to other levels of evidence in the determination of the level of therapeutic improvement and the selection of comparators.
7.3 Since uncertainty in the relative efficacy of a new drug product is common, high quality Level 1 evidence is preferred for new drug products to be assessed as having a breakthrough or substantial level of improvement relative to other drug products sold in Canada.
7.4 HDAP may consider lower levels of evidence, as required, on a case by case basis in order to determine comparative efficacy and toxicity or to assess the secondary factors which may impact the level of therapeutic improvement recommended by HDAP.
8. Selection of Comparable Medicines
8.1 HDAP uses the World Health Organization (WHO) Collaborating Centre for Drug Statistics Methodology's Anatomical Therapeutic Chemical Classification (ATC) System in the selection of comparable medicines.
8.2 Comparable medicines will typically be those identified under the ATC classification system at the sub-class level above the single chemical substance. This will normally be the fourth sub-class level. HDAP may also choose from the next higher sub-class or another sub-class. In some instances, it may be appropriate to select from the fifth or single chemical substance level.
8.3 HDAP may omit from the comparison a chemical substance or a drug product of the same ATC therapeutic class as the drug product under review if, in HDAP´s opinion, it is unsuitable for comparison. For example, drug products with primary indications/uses other than the primary indication/use of the drug product under review may be omitted from the comparison.
Procedures:
8.4 There will be no comparable medicines recommended by HDAP for a medicine that represents a breakthrough given that such a drug product is the first one to be sold in Canada that treats effectively a particular illness or condition.
8.5 For drugs that do not represent a breakthrough, HDAP will first attempt to identify medicines with the same approved indication or use, that are clinically equivalent to the product under review.
8.6 If no drugs are clinically equivalent in addressing the same approved indication or use of the drug under review, HDAP will identify all drug products that are either superior or inferior in treating the approved indication or use.
8.7 The comparable medicines for a new medicine that represents a new DIN of an existing dosage form of an existing medicine, or a new DIN of another dosage form of the medicine that is comparable to the existing dosage form will be limited to the same and/or different strength of the same active ingredient as the drug under review, unless the patentee has made a submission regarding therapeutic improvement.
8.8 The comparable medicines for a new combination product, where each of the elements of the combination product are sold in Canada, will be limited to the component parts, unless the patentee makes a submission regarding therapeutic improvement.
8.9 The comparable medicines for a new patented generic drug that is bioequivalent to a brand product sold in Canada, or that is a licensed version of the same brand product sold in Canada, will be limited to the same strength of the brand medicine.
Comparable Dosage Forms
8.10 For each comparable medicine identified, drug products of the same or comparable dosage form as the drug product under review will generally be selected. Schedule 3 lists the comparable dosage forms that HDAP uses to identify comparable drug products.
8.11 When comparable dosage forms cannot be identified, other dosage forms may be used if these dosage forms are comparable in addressing the approved indication or use.
8.12 Where evidence suggests that non-comparable dosage forms would be appropriate for comparison, these will be selected.
Comparable Dosage Regimens
8.13 The comparable dosage regimen recommended for comparison purposes will normally not be higher than the maximum of the usual recommended dosage in the Product Monograph (or similar information) taking into account relevant clinical variables. The most appropriate strength of the medicine will be chosen for a particular dosage regimen.
8.14 Generally, a dosage regimen based on a course of treatment will be applicable to acute indications, while a per-day regimen (based on maintenance dose) will be applicable to chronic situations.
9. Provisions for Over-the-Counter (OTC) and Veterinary Drug Products
9.1 As per the regulatory and reporting provisions outlined in Section I – Legal Framework, the scientific review for OTC and veterinary drug products will only be undertaken following the PMPRB's receipt of a complaint regarding the price of the drug product.
Procedures:
9.2 Upon receipt of a complaint, the PMPRB will undertake the scientific review of the OTC or veterinary drug product in the same manner as is undertaken for all other drug products, as outlined in this Chapter.
9.3 If a complaint is received for an OTC drug product, the required scientific information will be sent to HDAP to determine the level of therapeutic improvement, the comparable drug products, comparable dosage forms and comparable dosage regimens.
9.4 If a complaint is received for a veterinary drug product, a Veterinary Drug Advisory Panel (VDAP) will be formed to determine the level of therapeutic improvement, comparable drug products, comparable dosage forms and comparable dosage regimens.
Chapter 2 - The Price Review Process
1. Introduction
1.1 Upon completion of the Scientific Review Process, the Price Review Process is conducted for the purposes of:
- Establishing the MNE price for the drug product; and
- Assessing whether or not the price appears to be excessive.
2. Excessive Price Tests
2.1 The Therapeutic Class Comparison (TCC) Test compares the price of the drug product under review with the prices of the comparable drug products that are sold in the same markets at prices that the Board considers not to be excessive. The TCC test is described in Schedule 4. When a new drug product is a combination product, and either no submission by the patentee on therapeutic improvement has been made or when a submission by the patentee was made but HDAP has determined slight or no improvement over comparable drugs with the same primary approved indication or use, its price will be limited to the sum of each of the individual components.
2.2 The Reasonable Relationship (RR) Test considers the association between the strength and the price of the same medicine in the same or comparable dosage forms. The RR Test is described in Schedule 5. The RR Test is used when either no submission has been made by the patentee on therapeutic improvement or a submission by the patentee was made but HDAP has determined slight or no improvement over the same or different strength of the same or comparable dosage form of an existing drug product with the same approved indication or use as the drug product under review.
2.3 The Median International Price Comparison (MIPC) Test compares the ATP in Canada of the drug product under review with the median publicly available ex-factory prices of the same patented drug product sold in the countries listed in the Regulations. This test is described in Schedule 6. Note that if the new drug product is sold in less than five comparator countries, the MIPC test will be re-conducted once the drug is sold in five countries or after three years, whichever comes first.
2.4 The Highest International Price Comparison (HIPC) Test ensures that the drug product in Canada does not exceed the highest price at which the same drug product is sold in the comparator countries. This test is described in Schedule 6.
2.5 The International Therapeutic Class Comparison (ITCC) Test compares the ATP of the drug product with the publicly available ex-factory prices in the comparator countries listed in the Regulations of drug products identified in the domestic therapeutic class comparison. The ITCC test will only be conducted in cases of dispute regarding whether the price of the drug product under review appears to be excessive based on other price tests. It will not be used as a primary price test. This test is described in Schedule 7.
2.6 The CPI-Adjustment Methodology measures the change in the Consumer Price Index (CPI) over a specified period and is used to compare the ATP of a drug product with the CPI-adjusted price of the drug product. In cases where the ATP of a drug product declines from a previous year due to the provision of a benefit(s), as proven by evidence provided by the patentee, once the benefit ends if the patentee provides evidence that the price increase was solely due to the termination of the benefit, the MNE price of the drug product will be the previous highest non-excessive ATP (i.e., the De-Linking Methodology) instead of the MNE price resulting from the application of the CPI-Adjustment Methodology. The CPI-adjustment Methodology and the De-Linking Methodology are described in Schedule 8.
3. Review of Prices of New Patented Drug Products
3.1 Unless the exceptions in 2.1 or 2.2 apply, the test applicable to the introductory price of a new drug product is dependant on the level of therapeutic improvement recommended for the drug product during the Scientific Review Process (see Chapter 1).
Breakthrough:
The introductory price of a breakthrough new drug product will be presumed to be excessive if it exceeds the MIPC Test (Schedule 6).
Substantial Improvement:
The introductory price of a new drug product providing substantial improvement will be presumed to be excessive if it exceeds the higher of the prices of all inferior comparable drug products, based on a TCC Test (Schedule 4), and the MIPC Test (Schedule 6).
Moderate Improvement:
The introductory price of a new drug product providing moderate improvement will be presumed to be excessive if it exceeds the mid-point between the TCC Test of inferior drug products (Schedule 4) and the MIPC Test (Schedule 6) as long as the resulting MNE price is not less than the result of the TCC test, in which case the TCC test will determine the MNE price.
Slight or No Improvement:
The introductory price of a new drug product providing slight or no improvement will be presumed to be excessive if it exceeds the prices of all clinically equivalent drug products based on a TCC Test (Schedule 4).
3.2 Unless the patentee makes a submission claiming therapeutic improvement and HDAP identifies the drug product as providing moderate or substantial improvement,
- When a new drug product is a combination product, its price will be presumed to be excessive if it exceeds the sum of the prices of the individual components.
- The introductory price of a new drug product that is a new presentation of an existing dosage form of an existing medicine, or a new presentation of another dosage form of the existing medicine that is comparable to the existing dosage form, as per Schedule 3, will be presumed to be excessive if it exceeds the result of the RR Test (Schedule 5).
3.3 In addition to the tests above, the price of the new drug product will be presumed to be excessive if it exceeds the HIPC Test (Schedule 6).
3.4 Exceptions to the price tests outlined in 3.1 - 3.3 include:
- The introductory price of a new patented generic drug product that has been deemed by Health Canada to be bioequivalent to the reference brand drug product sold in Canada will be presumed excessive if it exceeds the price of the reference brand drug product. In this particular case, primary weight will be given to this modified TCC Test even if the Canadian price exceeds the price of the same drug product in the comparator countries.
- The introductory price of a new patented generic drug product that is a licensed version of a patented brand medicine will be presumed to be excessive if it exceeds the price of brand drug product sold in Canada.
- If the brand reference drug (under bioequivalence or licence) for the patented generic drug product is not sold in Canada, then an RR test will be conducted, unless the patentee makes a submission regarding therapeutic improvement. The HIPC test will also be conducted.
3.5 In addition to the price tests described above, the introductory prices for specific relevant markets will be reviewed to ensure they are not excessive.
Procedures:
3.6 The benchmark period is the period from the date of first sale to the end of the six-month regulatory reporting period (June 30 or December 31), as long as the period covered is greater than one month. If the period is less than one month, the following six-month reporting period will be used.
3.7 The introductory price of a new drug product is generally determined by calculating the ATP of the drug product during the benchmark period across all classes of customers in all provinces and territories.
3.8 Based on the application of the appropriate introductory price test(s) to first day of sale price and sales data, Board Staff will provide interim advice to the patentee as to whether or not the price would appear to be excessive.
3.9 The MNE price will only be established based on the benchmark period once the patentee files the price and sales data for that period.
3.10 Board Staff will calculate an ATP for each of the three classes of customers (hospital, pharmacy and wholesaler) to ensure that the price for each class of customer does not exceed the national MNE price for the drug product.
4. Review of Prices of Existing Patented Drug Products
4.1 The price of an existing drug product will generally be presumed to be excessive if it:
- exceeds the benchmark price of the drug product adjusted for the cumulative change in the CPI from the benchmark period to the pricing period under review, as per the CPI-Adjustment Methodology (Schedule 8), or
- exceeds the HIPC Test (Schedule 6).
4.2 An exception to 4.1 is when the ATP of a drug product declines from a previous year due to the provision of a benefit(s) and if once the benefit ends the patentee provides evidence to demonstrate that the price increase was due solely to the termination of the benefit. In this case, the MNE price will be the lower of the previous highest non-excessive ATP (i.e., the De-Linking Methodology in Schedule 8) and the HIPC test.
4.3 In addition, when a patentee can demonstrate that a national increase in the ATP is due solely to a sales-mix shift and none of the prices for each class of customer and in each province/territory exceed the MNE price as determined by the CPI-Adjustment Methodology, the ATP will be considered to not be excessive.
4.4 In the event that the actual change in the CPI is less than the forecast CPI and an apparent excessive price arises solely due to the patentee´s reliance on the forecast CPI, if the patentee reduces the price to comply with the MNE price based on the actual CPI in the next period, no excessive pricing will be presumed and no excess revenues will be calculated.
5. Existing Patented Drug Products Subsequently Sold by Another Patentee
5.1 Where an existing drug product is sold in Canada by a patentee other than the original patentee, the benchmark ATP of the drug product sold by the subsequent patentee cannot exceed the MNE price established in the last period the drug product was sold by the previous patentee.
5.2 The introductory benchmark price for the subsequent patentee is established as the lower of the MNE price of the previous patentee and the ATP of the subsequent patentee.
5.3 If the benchmark price of the subsequent patentee is set by the MNE price of the previous patentee, and where the subsequent patentee does not have access to the pricing information of the previous patentee, the CPI-Adjustment Methodology would be applied as though sales of that product were in the first year of sale.
5.4 If the benchmark price of the subsequent patentee is set by the MNE price of the previous patentee, and the subsequent patentee can demonstrate that they have access to the pricing information of the previous patentee, the CPI-Adjustment Methodology would be applied as though the new drug product was a continuation of the drug product of the original patentee. The subsequent patentee would therefore be allowed to keep any banked CPI price increases accrued by the original patentee, as per the CPI-Adjustment Methodology outlined in Schedule 8.
Chapter 3 – Investigations
1. Introduction
1.1 When the price of a patented drug product appears to exceed the Guidelines but not by an amount that triggers the investigation criteria (Schedule 9), the patentee will be expected to reduce the ATP so as to not be excessive and to offset any excess revenues that may have accrued, but no action will be taken by Board Staff.
1.2 When the price of a patented drug product appears to exceed the MNE price and the circumstances are within the criteria established by the Board (Schedule 9), Board Staff will conduct an investigation to determine the facts.
1.3 The examination will include an analysis of the pricing history of the drug product and where variability in average prices in different markets (i.e., hospital, pharmacy, wholesaler) appears to be an issue, Board Staff will conduct a price review at the level of the relevant market.
1.4 The period of time available to the patentee to respond to Board Staff following a notification that an investigation has been commenced is ordinarily brief. For example, if the patentee should have known that a price was outside the Guidelines based on its own filings (e.g., CPI-adjusted price), the period of time may be as short as seven calendar days. A longer period of time, 30 calendar days, may be available if it is reasonable to believe that the patentee might have been unaware that the ATP may appear to be excessive (e.g., if HDAP has recommended use of different comparators or dosage regimens from those which were proposed by and may have been reasonably anticipated by the patentee).
1.5 There are three possible outcomes to an investigation:
- The price is found to be within the Guidelines; or
- The price continues to exceed the Guidelines and the patentee submits an acceptable Voluntary Compliance Undertaking (VCU); or
- The price continues to exceed the Guidelines and the patentee does not submit an acceptable VCU in which case Board Staff will refer the matter to the Chairperson and recommend the issuance of a Notice of Hearing.
2. Within the Guidelines
2.1 If the investigation reveals that the price of the drug product was not outside the Guidelines, the investigation will be terminated and the patentee will be advised accordingly.
3. Voluntary Compliance Undertaking
3.1 If the investigation confirms that the price exceeded the Guidelines, the patentee will be given an opportunity to submit a written proposal in the form of a VCU to adjust its price and offset excess revenue accrued as a result of sales at an excessive price.
3.2 Board Staff will assist a patentee in preparation of a VCU, and may provide sample documents or other advice as may be appropriate to the situation.
3.3 If a patentee submits a VCU consistent with the Guidelines, it is the policy of the Board that only the Chairperson (or, if the VCU is submitted after the issuance of a Notice of Hearing, the Board Hearing Panel) may approve a VCU.
3.4 The Chairperson is not authorized to enter into negotiations on the terms of a VCU with a patentee.
3.5 The proposed VCU should include a statement as to the MNE prices with which the patentee agrees to comply and the means by which the patentee proposes to offset the excess revenues it received during the period the price was outside the Guidelines.
3.6 In most cases, the VCU should specify a payment to Her Majesty in Right of Canada as the means to offset excess revenues.
3.7 The proposal of a VCU does not constitute an admission by the patentee that the price of the drug product is or was excessive.
3.8 In deciding whether to accept a VCU, the Chairperson (or Board Hearing Panel) will be guided by section 83 of the Act and the policy of the Board that the price should be adjusted to conform to the Guidelines and that the patentee offset any excess revenues received since the price first exceeded the Guidelines.
3.9 The Board will report publicly on all VCUs accepted by the Chairperson or the Board. The information reported will ordinarily include the names of the drug product and the patentee and such other information as it considers appropriate. This information will be included in the PMPRB´s Annual Report and be published on the PMPRB Web site, and may also be published in the NEWSletter or other publications. Privileged or confidential information will not be published unless the information has been made public in a hearing.
4. Recommendation to Issue a Notice of Hearing
4.1 If the investigation confirms that the price exceeded the Guidelines, and the patentee did not submit an acceptable VCU, Board Staff will report the matter to the Chairperson with a recommendation that a Notice of Hearing be issued
Schedules
These Schedules are appended to the Guidelines in order to provide additional technical detail on a number of topics.
- Submissions by Patentees on Therapeutic Improvement
- Levels of Evidence for Determination of Therapeutic Improvement
- Comparable Dosage Forms
- Therapeutic Class Comparison Test
- Reasonable Relationship Test
- International Price Comparison Test
- International Therapeutic Class Comparison Test
- The CPI Adjustment and De-linking Methodology
- Criteria for Commencing an Investigation
Schedule 1 – Submissions by Patentees on Therapeutic Improvement
Each submission should clearly explain the rationale behind the patentee´s proposals for level of therapeutic improvement, for comparator drug products and for comparable dosage regimens.
The patentee should provide ten copies of the submission and all supporting references. Board Staff will verify that the supporting references mentioned or listed in the submission have been included and advise the patentee if any information is missing.
1. Supporting Clinical Evidence
1.1 Drug: name of drug, drug class, brief description of drug mechanism, approved indication(s) or uses, and approved or proposed dosing
1.2 Product Monograph (or similar information if no NOC): Submitted with Form 1, Identity of the Medicine
1.3 Individual Trials/Studies:
- Level 1 Evidence: Published randomized clinical trials (RCTs) of the drug under review versus active comparators, if any; Published RCTs of the drug under review versus placebo; High quality unpublished RCTs, if available.
- Published clinical trials with lower levels of evidence (e.g., outcome studies, systematic reviews of cohort and case-controlled trials) if Level 1 evidence is unavailable. Note: In relation to both Level 1 and other levels of evidence, the patentee is encouraged to focus the submission on key trials that lead to an NOC or to a change in clinical practice, or would be of the highest quality/best evidence the patentee has available.
- Editorials and errata of published clinical trials
- Other clinical evidence, such as ecological studies, case series and community surveys of the drug product under review if the patentee is proposing therapeutic improvements due to secondary factors.
1.4 Summary of trials included in submission in tabular format
- Study reference(s) (abstracts and publications if published), and study identification assigned by the patentee
- Brief description of the study and outcomes measures
- Trial Phase (i.e., Phase II, III or IV); Phase I trials will not be reviewed. 1.5 Brief overview of standards of therapy or accepted clinical practice for which the drug under review is indicated or used.
- For example, class reviews, systematic reviews/meta-analyses.
1.6 Published Clinical Practice Guidelines regarding the indication or use of the drug under review if available.
- Peer reviewed Canadian guidelines are preferred; American, UK, Australian and European guidelines will be considered.
2. Proposal of the Patentee
2.1 Executive Summary
- Brief description of the drug and its place in therapy, as well as a summary of the clinical evidence
2.2 Proposed level of therapeutic improvement
2.3 Proposed Comparators
- Evidence of the same approved indication or use as the drug product under review
2.4 Proposed comparable dosage regimens for the comparator and the drug product under review
- Approved or proposed doses
- Doses used in clinical trials
- Doses recommended in clinical practice guidelines
Schedule 2 – Levels of Evidence for Determination of Therapeutic Improvement 3
| Level |
Therapy/Prevention |
Economic and decision analyses |
| 1a |
SR (with homogeneity*) of RCTs |
SR (with homogeneity*) of Level 1 economic studies |
| 1b |
Individual RCT (with narrow Confidence Interval) |
Analysis based on clinically sensible costs or alternatives; systematic review(s) of the evidence; and including multi-way sensitivity analyses |
| 1c |
All or none§ |
Absolute better-value or worse-value analyses † |
| 2a |
SR (with homogeneity*) of cohort studies |
SR (with homogeneity*) of Level >2 economic studies |
| 2b |
Individual cohort study (including low quality RCT; e.g., <80% follow-up) |
Analysis based on clinically sensible costs or alternatives; limited review(s) of the evidence, or single studies; and including multi-way sensitivity analyses |
| 2c |
"Outcomes" Research; Ecological studies |
Audit or outcomes research |
| 3a |
SR (with homogeneity*) of case-control studies |
SR (with homogeneity*) of 3b and better studies |
| 3b |
Individual Case-Control Study |
Analysis based on limited alternatives or costs, poor quality estimates of data, but including sensitivity analyses incorporating clinically sensible variations. |
| 4 |
Case-series (and poor quality cohort and case-control studies§§) |
Analysis with no sensitivity analysis |
| 5 |
Expert opinion without explicit critical appraisal, or based on physiology, bench research or "first principles" |
Expert opinion without explicit critical appraisal, or based on economic theory or “first principles“ |
| * |
Homogeneity means a systematic review that is free of worrisome variations (heterogeneity) in the directions and degrees of
results between individual studies. Not all systematic reviews with statistically significant heterogeneity need be worrisome,
and not all worrisome heterogeneity need be statistically significant. As noted above, studies displaying worrisome heterogeneity
should be tagged with a "-" at the end of their designated level.
|
| § |
Met when all patients died before the Rx became available, but some now survive on it; or when some patients died before
the Rx became available, but none now die on it.
|
| §§ |
Poor quality cohort study is defined as a study that failed to clearly define comparison groups and/or failed to measure exposures
and outcomes in the same (preferably blinded), objective way in both exposed and non-exposed individuals and/or failed
to identify or appropriately control known confounders and/or failed to carry out a sufficiently long and complete follow-up of
patients. A poor quality case-control study is defined as a study that failed to clearly define comparison groups and/or failed
to measure exposures and outcomes in the same (preferably blinded), objective way in both cases and controls and/or
failed to identify or appropriately control known confounders.
|
| † |
Better-value treatments are clearly as good but cheaper, or better at the same or reduced cost. Worse-value treatments are as
good and more expensive, or worse and equally or more expensive. |
Schedule 3 – Comparable Dosage Forms
This Schedule identifies comparable dosage forms for the purpose of conducting therapeutic class comparisons and TCC and RR price tests for new drug products. Formulations within each class are considered comparable, but dosage forms in a different class are not.
The PMPRB reviews the list of comparable dosage forms periodically to ensure that it includes those currently used.
| Comparable Dosage Forms |
| Topical |
Nasal/Pulmonary |
Oral Solid |
Aerosol
Cream
Gel
Liquid
Ointment
Paste
Powder
Shampoo
Spray
Patches
Disks
Dressings |
Drops
Aerosol
Spray
Solution
Powder
Gas
Metered dose preparations |
Tablet
Capsule
Modified release tablets
Modified release capsules
Effervescent powder
Effervescent tablets
Effervescent granules
Caplet
Modified release caplets |
| Oral Liquid |
Vaginal |
Parenteral |
Solution
Powder for solution
Powder for suspension
Suspension
Drops
Modified release liquid |
Suppository
Cream
Tablet
Douche
Foam
Cone
Ovule
Gel
Tampon
Sponge
Insert |
Solution
Powder for solution
Suspensions or emulsions
Modified release injections
Implant |
| Optic/Ophthalmic |
Rectal |
Dental/Sublingual Buccal |
Liquid
Powder for solution
Drops
Suspension
Ointment
Gel
Modified release ocular devices |
Suppository
Cream
Ointment
Enema
Suspension
Foam |
Mouth wash
Solution
Suspension
Powder for suspension
Lozenge
Gel
Gum
Modified release buccal tablets
Sprays - sublingual
Sprays - buccal
Sublingual tablets
Tooth paste
Tooth powder |
Schedule 4 – Therapeutic Class Comparison Test
1. Approach
The Therapeutic Class Comparison (TCC) Test compares the price of the drug product under review with the price of the comparable drug products sold in the same market at prices that the PMPRB considers not to be excessive. Comparable drug products are first selected and then their prices are compared against the drug product under review.
2. Selection of Comparable Drug Products
The identification of comparable drug products is a function of the scientific review process. The selection is comprised of two elements:
- the identification of comparable medicines; and
- the identification of comparable dosage forms.
For complete details on the selection of comparable drug products, please refer to Section III, Chapter 1.
3. Measuring the Price
The PMPRB considers it appropriate to compare the prices of comparable drug products taking into consideration the dosage regimens and other clinically relevant variables required to produce a clinically equivalent effect. The PMPRB will make these price comparisons in terms of the price per course of treatment or price per day, whichever is more applicable. Generally, the price per course of treatment will be applicable to acute indications, whereas price per day (based on maintenance dose) will be applicable to chronic situations.
The dosage regimens for both the drug product under review and the comparable drug products to be used in the TCC Test are recommended by HDAP and by Board Staff. For further details on this process, please refer to Section III, Chapter 1.
For comparison purposes, Board Staff will use an appropriate public source for the price of the comparable drug products. The appropriateness of the prices used in the TCC Test will be determined on a case-by-case basis.
Board Staff reserves the right to exclude from the TCC Test any drug product it has reason to believe is being sold at an excessive price.
When an identified comparable drug product is patented and sold by the same patentee as the drug product under review, the comparable drug product's average transaction price based on the patentee´s submission to the PMPRB (or, if outside the Guidelines, its MNE price) may be used for the comparison.
The highest non-excessive price of the comparators will be used to establish the MNE price for the new drug product.
The Board has identified a De-linking Methodology for existing drug products whose prices drop below a previous non-excessive ATP (see Schedule 8). Normally, the patentee would only need to provide evidence when the benefit ends, that the subsequent price increase was due to the end of a benefit in order to take advantage of the ability of the price of the drug product to “rebound” to its previous non-excessive ATP.
However, the patentee may choose to ask the PMPRB to publish the previous higher ATP as the drug product´s MNE price while the benefit is still in effect in order that, for the purposes of a TCC Test for a new drug product for which the existing drug product is likely to be a comparator, the price used in the TCC test for the new drug product would be the previous higher non-excessive ATP of the existing comparator and not the actual ATP of the existing comparator that has dropped due to the provision of a benefit. If the patentee chooses to ask the PMPRB to publish the former higher non-excessive ATP as the MNE price for the existing drug, then this price will be used in all cases where the existing drug is an appropriate comparator, whether the new drug is sold by the same patentee or another patentee.
Note: This Guideline will only apply if the price of the new drug product does not also reflect a reduced price due to a benefit.
Schedule 5 – Reasonable Relationship Test
Reasonable relationship refers to the association between a new drug product and existing forms of the same medicine of the same or different strength sold in same or comparable dosage forms. The reasonable relationship test defines a maximum non-excessive (MNE) introductory price for the new drug product. This schedule describes in general terms the process by which reasonable relationship may be determined.
The determination of reasonable relationship will be based on one of three possible tests. Only one test will be appropriate for a particular new drug product.
To determine which test is appropriate for a particular case, the three are considered in descending order.
Test 1: Same Strength Test
If there are one or more comparable drug products of the same strength as the new drug product, then the highest priced comparable drug product of the same strength determines the MNE price for the new drug product. Prices above this threshold are considered to be outside the PMPRB´s Guidelines. The result of this test takes precedence over the other two tests, regardless of their outcomes.
In Figure 1 below, given three comparators of equal strength but different prices (P1, P2, and P3) a new drug will have an MNE price equal to that of the highest priced comparator, in this case P1.
Figure 1 - Same Strength Test

Test 2: Unit Price Linear Relationship Test
If it is not possible to conduct Test 1 (Same Strength Test), and there are two or more comparable drug products, this test will be conducted.
The test is conducted in a series of steps:
- As shown in Figure 2A below, lines are drawn for all possible pairs of comparable drug products (e.g., A to B, A to C, B to C).
- The pair with a positive slope and highest Y-axis intercept determines the Y-intercept that will be used to determine the MNE price line threshold. In the example in Figure 2A, the highest Y-intercept results from the line running from A to B.
Figure 2A - Linear Relationship Test - Representing Steps 1-2
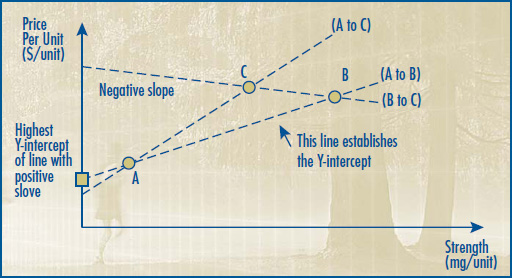
The MNE price line threshold is produced using the Y-intercept resulting from Step 2, and the point representing the per unit price of the highest priced comparable drug product. In the example in Figure 2B, the comparable drug product C has the highest price per unit. A line is then drawn between point C and the previously determined Y-intercept.
- Prices of new comparable drug products will not be excessive if they do not exceed the MNE price line threshold.
Figure 2B - Linear Relationship Test - Representing Steps 3-4
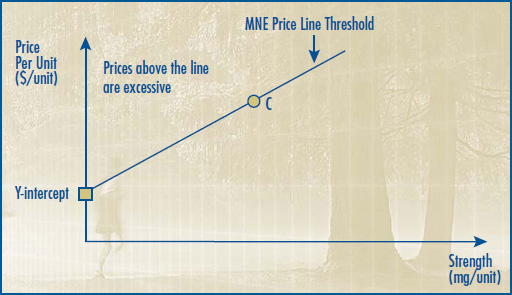
The result of this test takes precedence over Test 3, regardless of the result of that third test.
Test 3: Different Strength Test
This test is used when there is only one other (higher or lower) strength of a comparable drug product sold in Canada. Although there is only one other strength sold, there could be several products of this strength sold. The price of the new drug product under review is considered in relation to the highest price of the comparable drug product of the different strength.
The MNE price is determined by calculating the ratio of the strength of the new drug product over the strength of the existing drug product and multiplying this ratio by the price of the existing drug product.
For Example:
- A 5 mg strength drug product is being sold and the highest price at which it is sold is $10.
A 7.5 mg strength drug product of the same medicine in a comparable dosage form is introduced.
The price of the new 7.5 mg drug product cannot exceed $15.00.
7.5mg/5mg x $10 = $15
Schedule 6 – International Price Comparison Test
1. Median International Price Comparison (MIPC) Test
1.1 The price of the drug product under review will be compared with the median of the ex-factory prices of the same strength and dosage form of the same patented medicine for each country listed in the Regulations (France, Germany, Italy, Sweden, Switzerland, the United Kingdom, and the United States).
1.2 When the International Price Comparison is being conducted to determine the median price and the drug product is sold in an even number of countries, the median will generally be the simple average of the middle two prices.
1.3 When the International Price Comparison is being conducted to determine the median price, an interim median international price will be used in cases when the medicine is sold in fewer than five countries at the time of its introduction. Unless it is excessive, the introductory price will be treated as the interim benchmark price. The interim benchmark price may be reviewed at the end of three years or when the same patented medicine, strength and dosage form is sold in at least five countries, whichever comes first.
2. Highest International Price Comparison (HIPC) Test
2.1 For both new and existing drug products, the price at which the drug product is sold in Canada cannot exceed the prices in which it is sold in countries listed in the Regulations.
2.2 Where the price of a drug product in Canada is higher than the international prices for the same drug product, it will not be forced down to a price that is below that of the same or comparable dosage form that is of the same or lower strength.
3. Indirect International Price Comparison
3.1 When a direct international price comparison of the drug product under review is not possible because the drug product is only sold in Canada, the most similar strengths of comparable dosage forms as per Schedule 3 will be considered for each of the comparator countries.
4. Exchange Rates
4.1 In order to apply the highest of the International Price Comparison outlined in Section III, Chapter 2, the exchange rates used are ordinarily the simple average of the thirty-six monthly average noon spot exchange rates for each country (taken to eight decimal places), as published by the Bank of Canada for the thirty-six months ending with the last month of the pricing period under review.
e.g., The pricing period under review is July to December 2007. The exchange rates used, for the purposes of the highestprice Guideline, are for the months of January 2005 through December 2007.
4.2 To review the introductory price of a new drug product, the exchange rates used are the simple average of the thirty-six monthly average noon spot exchange rates for each country (taken to eight decimal places) as published by the Bank of Canada for the thirty-six months ending four months before the date of the first sale of the drug product.
e.g., If the new drug product under review was first sold in October 2008, the exchange rates used are for the months of June 2005 through May 2008.
4.3 Exchange rates are published on the PMPRB web site on a monthly basis, under Frequently Requested Items.
5. Existing Medicines with Unusual Circumstances
5.1 The Guidelines require that patentees take appropriate action when an investigation verifies that its price exceeds the Guidelines. There are, however, circumstances where a product whose price is within the Guidelines in one review period is then found to be outside the Guidelines, as a result of the application of the highest of the International Price Comparison, in a subsequent period as a result of events other than actions directly attributed to the patentee. The following are examples of three such circumstances:
- Exchange rate variations;
- A foreign regulator forcing price reductions; or
- A drug product priced highest in the world is removed from the market.
Under the circumstances identified above, patentees will be notified that their price is outside the Guidelines and will be expected to adjust the product´s price by the next year to be within the Guidelines or be subject to a Voluntary Compliance Undertaking (VCU) and repayment of excess revenues dating back to the original excessive price.
Schedule 7 – International Therapeutic Class Comparison Test
1. Concept and Application
1.1 The International Therapeutic Class Comparison (ITCC) compares the price of the drug product under review with the prices of comparable drug products that are sold in the seven comparator countries listed in the Regulations: France, Germany, Italy, Sweden, Switzerland, the United Kingdom, and the United States.
1.2 This test is not a primary price test. However, it may provide useful information in the context of an investigation into apparent excessive prices. This price test will also be performed by Board Staff for the information of a Board Panel in the case of a Hearing.
2. Selection of Comparable Drug Products
2.1 For the purpose of the ITCC test, the comparable drug products identified in the domestic TCC test will be used. For more details on the selection of comparator drug products for the TCC test, please refer to Section III, Chapter 1.
3. Derivation of the ITCC Test
3.1 The following two variations may be used to calculate the ITCC test:
- The ratio of the price of the drug product under review relative to the prices of comparable drug products in the countries listed in the Regulations is calculated. This ratio is then applied to the price of the drug product under review in Canada.
- The prices of all of the comparable drug products in the comparator countries are identified and a series of statistical values are calculated: mean, median, and range (interval between the maximum and minimum price).
3.2 Where a price of a comparable drug product was excluded from the TCC test, because it appeared to be excessive, it will also be excluded from the conduct of the ITCC test.
4. Exchange Rates
4.1 The exchange rates used are ordinarily the simple average of the thirty-six monthly average noon spot exchange rates for each country (taken to eight decimal places), as published by the Bank of Canada for the thirty-six months ending with the last month of the pricing period under review. e.g., The pricing period under review is July to December 2007. The exchange rates used are for the months of January 2005 through December 2007.
4.2 In regard to the introductory price of a new drug product, the exchange rates used are the simple average of the thirty-six monthly average noon spot exchange rates for each country (taken to eight decimal places), as published by the bank of Canada for the thirty-six months ending four months before the date of the first sale of the drug product.
e.g., If the new drug product under review was first sold in October 2008, the exchange rates used are for the months of June 2005 through May 2008.
4.3 Exchange rates are published on the PMPRB Web site on a monthly basis, under Frequently Requested Items.
Schedule 8 – CPI-Adjustment and De-linking Methodology
1. The Concept
1.1 The price of an existing drug product during the year under review will be presumed to be excessive if it exceeds the benchmark price of the drug product adjusted for the three year cumulative change in the Consumer Price Index (CPI) from the benchmark year to the year under review (CPI-adjusted price).
1.2 In addition, the price increase in any one year may not exceed 1.5 times the forecast change in the annual CPI.
1.3 In periods of high inflation (over 10%), the limit will be 5 percentage points more than the forecast change in the annual CPI.
2. Terminology
2.1 Forecast Period:
The forecast period is the year for which prices are being set.
2.2 Introductory Period:
The introductory period for new drug products is the period from the date of first sale to the end of the six-month regulatory reporting period (i.e., June 30 or December 31) when that period is greater than one month. For example, a drug product first sold in March 2007 would have an introductory period of March to June 2007, whereas a drug product first sold in December 2007 would have an introductory period of January to June 2008.
2.3 Benchmark year:
- For patented drug products first sold in Canada more than three years prior to the forecast period, the benchmark year is the calendar year three years proceeding the forecast period. For example, for 2009 the corresponding benchmark year is 2006.
- For patented drug products first sold three years or less prior to the forecast period, the benchmark year is the year in which the drug product was first sold in Canada.
2.4 Benchmark Price:
- For patented drug products first sold in Canada more than three years prior to the forecast period, the benchmark price of the drug product is its ATP three years prior to the forecast period based on the patentee's Form 2, Block 4 submission, or if that price was outside the Guidelines, the MNE price, in the benchmark year.
- For patented drug products first sold three years or less prior to the forecast period, the benchmark price of the drug product is its ATP in the introductory period based on the patentee's Form 2, Block 4 submission, or if that price was outside the Guidelines, the MNE price, during its introductory period. This benchmark price establishes the MNE price for the benchmark year.
- As described below in Paragraph 3, under specific, limited circumstances, the MNE price may be determined using the De-Linking Methodology in place of the CPI-Adjustment Methodology.
2.5 Base CPI:
The average of the monthly increases (seasonally adjusted at an annual rate) in the CPI, as published by Statistics Canada, for the benchmark year. The base CPI figures are calculated and published annually by the PMPRB in April.
2.6 Forecast CPI:
The forecast of the CPI increase relevant to the forecast period is based on the previous year's actual CPI published by Statistics Canada and adjusted for the latest annual inflation projections by the federal Department of Finance. The PMPRB's forecast CPI is calculated prior to the beginning of the forecast period and published annually in April.
2.7 CPI-Adjustment Factor:
The forecast CPI divided by the base CPI, rounded to three decimal places.
2.8 CPI-Adjusted Price:
This is the benchmark price multiplied by the CPI-adjustment factor.
2.9 Cap:
One year price increases may not exceed 1.5 times the forecast change in the annual CPI. In times of high inflation (greater than 10%), the limit will be 5 percentage points more than the forecast change in the CPI.
2.10 Example of the application of the CPI-Adjustment Methodology:
Forecast Period: Jan – Dec 2009
First sale: 1998
Benchmark Year: 2006
Benchmark Price: $10.00
Price in 2008: $10.39
CPI-adjusted price: 1.065 (CPI-adjustment factor) × $10.00 = $10.65
Cap: 1.030 (1.5 x Forecast CPI) × $ 10.39 = $10.70
The MNE price for the drug product is $10.65
3. De-Linking Methodology
3.1 Defining “De-Linking”
Under the CPI-Adjustment Methodology, the MNE price is “linked” to the ATP of the three previous years. However, in certain specific circumstances and with necessary evidence from the patentee, the MNE price may be “de-linked” from the ATP of the previous three years such that it may increase up to the previous highest non-excessive ATP.
3.2 When is De-Linking Available to Patentees?
The De-Linking Methodology is available when the national ATP declines from a previous higher non-excessive ATP, due to a benefit. The De-linking Methodology is only applied at the time when the benefit ends and only if the patentee submits evidence to the PMPRB that shows the apparently excessive increase in the ATP was due solely to the termination of a benefit. The De-linking Methodology ceases to be available to the patentee once the ATP reaches or exceeds the previous highest non-excessive ATP.
3.3 Defining Benefits
For the purpose of calculating the ATP, benefits include price reduction given as a promotion or in the form of rebates, discounts, refunds, free goods, free services, gifts or any other benefits of a like nature that are connected to a sales transaction and after deduction of the federal sales tax.
3.4 Evidence of Benefits Required
Patentees wishing to use the De-Linking Methodology are required to provide evidence of the cancellation or reduction in uptake of a benefit. The exact form of this evidence (i.e., a contract), the type of data (i.e., quantity, price, and revenue) and timing of this evidence will depend on the specifics of each case. A patentee will not be allowed to take advantage of the De-Linking Methodology if the apparent reduction in the national ATP is due solely to new sales to a new class of customer at a lower price.
3.5 Example of the application of the CPI-Adjustment Methodology where a drug product is subject to de-linking
Previous highest non-excessive ATP: $10.00
Price in 2007: $8.00
Price in 2008: $10.00
If evidence is submitted by the patentee that demonstrates a previous benefit was no longer in effect in 2008, the MNE price would be determined by the previous highest non-excessive ATP (no matter in what year the drug product was sold at that price), and therefore the price in 2008 would be considered not excessive.
Schedule 9 – Criteria for Commencing an Investigation
The Guidelines provide that the Board may establish criteria for the commencement of an investigation into an apparent excessive price.
The criteria balance the need for pricing flexibility on the part of patentees with the PMPRB's mandate of protecting consumers by ensuring that the prices of patented drug products are not excessive. The Board publishes its criteria for commencing an investigation to improve transparency and to provide patentees with greater certainty as to their responsibilities.
A price is considered to be within the Guidelines if the price is equal to or below the MNE price. A price is considered to appear excessive if the price exceeds the MNE price.
The criteria for commencing an investigation represent the standards the Board applies in order to allocate its resources to investigations as efficiently as possible. Their existence should not be construed as indicating that the Board accepts any deviation from the Guidelines. The Board is satisfied that its criteria ensure that all significant cases of pricing outside the Guidelines will be subject to an investigation.
Where a price exceeds the MNE price by an amount too small to trigger an investigation in one year, the patentee is expected to reduce the price in the following year so as to be below the MNE price and to offset any excess revenues. However, cumulative excess revenues cannot fall below zero. Evidence of persistent pricing outside the Guidelines, even by an amount which does not trigger the investigation criteria, may result in commencing an investigation.
Should the price of a patented drug product or its cumulative excess revenues ever meet the criteria, an investigation will be initiated in conformity with the Guidelines. Patentees will be advised of cumulative excess revenues for each of their drug products as part of the compliance reports they receive from the PMPRB.
| Criteria for Commencing an Investigation |
|
Board Staff will commence an investigation into the price of a patented drug product when any of the following criteria are met:
- The ATP is more than 5% above the MNE price.
- Excess revenues are $50,000 or more.
- PMPRB receives a complaint that a price is excessive.
|
1 ICN Pharmaceuticals, Inc. v. Canada (Staff of Patented Medicine Prices Review Board) (C.A.) [1997] 1 F.C. 32, aff´g [1996] F.C.J. No. 206 (T.D.); see also Hoechst Marion Roussel Canada Inc. v. Canada (Attorney General) [2005] F.C.J. No. 1928 (T.D.)
2 Shire BioChem Inc. v. Attorney General of Canada [2007] FC 1316
3 The table above is based on the Oxford Centre for Evidence-based Medicine Levels of Evidence (May 2001) - produced by Bob Phillips, Chris Ball, Dave Sackett, Doug Badenoch, Sharon Straus, Brian Haynes, Martin Dawes since November 1998.
SR: Systematic Review
RCT: Randomized Clinical Trials
Rx: Therapy