Compendium des politiques, des Lignes directrices et des procédures
Table des matières
- Introduction et objectif de la consultation auprès des intervenants
- Positions du Conseil concernant les modifications proposéesà son Compendium des politiques, des Lignes directriceset des procédures
- Préambule
- Partie I – Le cadre juridique
- Partie II – Les politiques
- Partie III – Lignes directrices et procédures
- Chapitre 1 – Le processus d’examen scientifique
- Chapitre 2 – Le processus d’examen du prix
- Chapitre 3 – Enquêtes
- Appendices
- Appendice 1 – Proposition du breveté sur le niveau d’amélioration thérapeutique
- Appendice 2 – Formes posologiques comparables
- Appendice 3 – Comparaison selon la catégorie thérapeutique
- Appendice 4 – Test de la relation raisonnable
- Appendice 5 – Test de la médiane des prix internationaux
- Appendice 6 – Comparaison du prix au Canada avec le prix international le plus élevé
- Appendice 7 – Comparaison du prix au Canada selon la catégorie thérapeutique internationale
- Appendice 8 – Tests appliqués aux prix des nouveaux produits médicamenteux
- Appendice 9 – Méthodologie de rajustement du prix selon l’IPC
- Appendice 10 – Méthodologie de la majoration
- Appendice 11 – Critères justifiant la tenue d’une enquête
- Appendice 12 – Examens du prix « sur un marché »
- Appendice 13 – Remboursement des recettes excessives
Introduction et objectif de la consultation auprès des intervenants
Le Conseil d’examen du prix des médicaments brevetés a préparé une autre version de son Compendium des politiques, des Lignes directrices et des procédures (Compendium) aux fins de la quatrième phase de sa révision de ses Lignes directrices. La présente ébauche se situe dans le prolongement des différentes activités menées depuis le début du processus de révision des Lignes directrices en 2005, et plus particulièrement depuis la publication du document d’Avis et commentaires le 20 août 2008.
En application du paragraphe 96(5) de la Loi sur les brevets, le Conseil consulte ses intervenants sur cette dernière ébauche du Compendium pour mieux comprendre leurs points de vue sur les modifications proposées à ses politiques, ses lignes directrices et ses procédures. Le présent document de consultation est composé des deux parties suivantes :
- Partie A – Sommaire des questions soumises à la consultation ainsi que des points de vue formulés par les intervenants depuis la publication du dernier document d’Avis et commentaires (août 2008) ainsi que les positions du Conseil concernant ces questions, et
- Partie B – Ébauche révisée du Compendium des politiques, des lignes directrices et des procédures.
L’objectif de la présente consultation est de recueillir vos points de vue concernant les dernières modifications apportées à l’ébauche révisée du Compendium depuis la version qui vous a été soumise le 20 août 2008. D’autres sections du Compendium, celles-là nommées dans la partie A, ont été approuvées par le Conseil dans la foulée des consultations antérieures et ne sont donc pas soumises à nouveau à vos commentaires.
Le Conseil prendra connaissance de tous les mémoires qui lui seront transmis et publiera la version finale du Compendium révisé au début de juin 2009. Le nouveau Compendium devrait entrer en vigueur le 1er juillet 2009.
Nous vous saurions gré de bien vouloir faire parvenir vos mémoires directement à la Secrétaire du Conseil, Mme Sylvie Dupont, d’ici au 27 avril prochain à l’adresse électronique suivante :
sylvie.dupont@pmprb-cepmb.gc.ca
ou encore à l’adresse postale suivante :
BoîteL40
Centre Standard Life
333, avenue Laurier ouest
Bureau 1400
Ottawa (Ontario) K1P 1C1
Si votre mémoire est volumineux, nous vous saurions gré de joindre à celui-ci un sommaire dans lequel sont exposés les principaux points traités dans votre mémoire. Par souci d’ouverture et de transparence, le Conseil affichera dans son site Web tous les mémoires qui lui auront été soumis dans le cadre de la présente consultation.
Positions du Conseil concernant les modifications proposéesà son Compendium des politiques, des Lignes directriceset des procédures
Le présent document donne un aperçu des principaux points d’achoppement sous examen, dont ceux soulevés dans le cours de la consultation en août dernier sur les premières modifications proposées aux Lignes directrices. Dans le présent document, le contexte dans lequel s’inscrit chaque point est discuté et les points de vue exprimés par les intervenants l’automne dernier sont rapportés après quoi sont présentés la position du Conseil et (ou) le suivi qu’il envisage donner.
Changements apportés au Compendium du Conseil
Dans l’ébauche du Compendium révisé soumis à la consultation en août dernier, le Conseil a proposé plusieurs changements, nommément des modifications à la structure générale du Compendium afin d’améliorer la circulation de l’information, l’ajout de la section « Cadre juridique » afin de permettre une meilleure compréhension des intervenants et la révision du libellé du mandat afin de mieux refléter la raison d’être du CEPMB.
Points de vue des intervenants
D’une façon générale, les intervenants ont bien accueilli la nouvelle structure du Compendium ainsi que la précision faite relativement au cadre juridique du CEPMB. Les intervenants ont toutefois exprimé des opinions divergentes concernant le libellé du mandat du Conseil. Pour certains intervenants, la raison d’être du CEPMB est de veiller à ce que les brevetés ne vendent pas leurs médicaments brevetés à des prix excessifs au Canada afin de protéger les intérêts des consommateurs et de contribuer au régime canadien de soins de santé alors que pour d’autres le Conseil devrait avoir pour mandat d’assurer un juste équilibre entre l’encouragement de la recherche-développement pharmaceutique au Canada et la protection contre la vente de médicaments à des prix excessifs.
Position du Conseil
De l’avis du Conseil, le CEPMB a été créé en 1987 pour protéger les intérêts des consommateurs canadiens et pour contribuer au régime de soins de santé. Le Conseil a donc décidé d’exprimer clairement cet objectif dans le libellé révisé de son mandat.
De plus, dans le document d’Avis et commentaires d’août 2008, le Conseil écrivait dans la section 8 du « Cadre juridique » que tout élément d’information que le breveté soumet au CEPMB qui appartient déjà au domaine public ne sera pas considéré confidentiel en vertu du paragraphe 87(1) de la Loi sur les brevets (Loi). Même si le Conseil n’a pas l’habitude de publier les prix départ-usine accessibles au grand public d’un médicament breveté au Canada et dans les pays de comparaison nommés dans le Règlement sur les médicaments brevetés (formulaire 2, section 5) sans avoir obtenu au préalable l’autorisation du breveté, le Conseil estime qu’il n’y a pas lieu de continuer de considérer confidentiels les renseignements fournis dans la section 5 du formulaire 2 étant donné qu’ils sont par définition accessibles au grand public.
| Sommaire des principaux changements soumis à la consultation |
Emplacement dans le Compendium révisé |
| Le libellé du mandat du CEPMB a été modifié pour se lire comme suit : « veiller à ce que les prix auxquels les brevetés vendent leurs médicaments brevetés au Canada ne soient pas excessifs conformément aux intérêts des consommateurs et du régime de soins de santé ». |
Partie I, paragraphe 2.1 |
| La section « Cadre juridique » du Compendium a été révisée de manière à inclure expressément dans la définition les éléments d’information accessibles au grand public les prix de vente des produits médicamenteux brevetés dans les pays de comparaison dont le breveté fait rapport au CEPMB (formulaire 2, section 5). |
Partie I, paragraphe 9.2 |
Remplacement de l’expression « Prix maximum non excessif »
Dans le cours de discussions récentes, des intervenants de l’industrie ont exprimé l’opinion que l’expression utilisée pour décrire le prix maximum autorisé donne la fausse impression qu’il s’agit d’un prix plafond qui ne peut être dépassé.
Points de vue des intervenants
Les intervenants représentant l’industrie estiment que l’expression « prix maximum non excessif » donne à penser qu’il s’agit d’un prix plafond qui ne peut être dépassé. Toutefois, le CEPMB fait l’examen du prix moyen des produits médicamenteux brevetés sur un marché. Étant donné que les prix des produits médicamenteux peuvent varier selon le marché, l’expression « prix de transaction moyen national » peut laisser sous-entendre une certaine variabilité des prix. Tant que le prix du produit médicamenteux sur chaque marché ne dépasse pas le Prix maximum non excessif établi pour la période de lancement et, pour les périodes subséquentes, n’augmente pas dans une mesure plus grande que ne le permet la méthodologie du prix rajusté selon l’IPC ou n’est pas plus élevé au Canada que dans tous les pays de comparaison, le prix du produit médicamenteux ne sera pas considéré excessif. Toutefois, lorsque comparé au Prix de transaction moyen national et au Prix maximal non excessif national, le Prix de transaction moyen du marché peut sembler excessif.
Position du Conseil
Le Conseil est lui aussi d’avis qu’il est important que la terminologie qu’il utilise ne porte pas à confusion afin d’être aussi transparent que possible pour ses intervenants. Le Conseil estime qu’il est également nécessaire de réviser la terminologie afin que les intervenants comprennent encore mieux la mécanique de révision du prix au niveau des différents marchés. Ainsi donc, le Conseil sollicite les points de vue de ses intervenants concernant la pertinence des expressions suivantes :
- L’expression « Prix maximal non excessif pour la période de lancement » serait remplacée par « Prix moyen maximal potentiel », indiquant ainsi que ce prix établira la limite supérieure du prix moyen sur les différents marchés (national, catégorie de clients, province/territoire) pour la période de lancement.
- Dans le cas des produits médicamenteux existants, l’expression « Prix maximal non excessif » serait remplacée par « Prix moyen non excessif ». Ainsi, chaque marché (national, catégorie de clients, province/territoire) aurait son propre prix moyen non excessif. Ce prix serait calculé à l’aide des prix de transaction moyens non excessifs réels du marché des années antérieures.
| Sommaire des principaux changements soumis à votre consultation |
Emplacement dans le Compendium révisé |
| Dans le cas des nouveaux produits médicamenteux, l’expression « Prix moyen maximal potentiel » serait utilisée au lieu de « Prix maximum non excessif pour la période de lancement ». |
Dans l’ensemble des Lignes directrices |
| Dans le cas des produits médicamenteux existants, l’expression « Prix moyen non excessif » serait utilisée au lieu de Prix maximum non excessif ». |
Dans l’ensemble des Lignes directrices |
Publication du Prix moyen maximal potentiel rajusté selon l’IPC
Les intervenants représentant l’industrie ont demandé au Conseil de publier le prix moyen maximal potentiel (qui correspond à la limite supérieure des prix sur tout marché pour la période de lancement) ainsi que ce prix rajusté annuellement selon l’IPC.
Le Conseil ne s’oppose pas à cette proposition des intervenants représentant l’industrie, mais avant de l’appliquer sollicite les commentaires des intervenants dans le cadre de la présente ronde de consultations.
Il convient de préciser que le Prix moyen maximal potentiel rajusté selon le taux de l’IPC n’est pas utilisé pour les fins de la réglementation. On ne peut en effet assumer que le Prix moyen maximal potentiel rajusté selon le taux de l’IPC représente un prix moyen non excessif pour un marché donné. Le Prix moyen non excessif des produits médicamenteux existants continuera d’être établi à l’aide des Prix de transaction moyens réels de chaque marché, des augmentations selon l’IPC autorisées ainsi que du Prix international le plus élevé.
Il convient aussi de préciser que les Prix moyens maximaux potentiels rajustés selon le taux de l’IPC ne constituent pas des prix publics qui peuvent être utilisés aux fins de l’examen du prix qu’effectue le CEPMB.
Enfin, les dispositions de la Loi concernant le caractère confidentiel des renseignements déposés par les brevetés prévoient que tout prix publié doit provenir d’une source publique par opposition aux prix confidentiels dont les brevetés doivent faire rapport au Conseil. Il n’est pas toujours possible pour le CEPMB de trouver des prix publics qui sont convenables aux fins de rapport et il arrive qu’il n’en trouve pas.
Niveaux d’amélioration thérapeutique
Au cours des consultations menées en 2006, les intervenants ont exprimé certaines réserves relativement à l’approche du CEPMB en ce qui a trait à l’évaluation du niveau d’amélioration thérapeutique d’un produit médicamenteux. Selon ces intervenants, l’approche ne reconnaîtrait pas la nature des innovations pharmaceutiques progressives. D’autres consultations ont été tenues auprès des intervenants et un Groupe de travail sur les améliorations thérapeutique a été créé et chargé de formuler des recommandations sur la façon d’améliorer le mode d’évaluation du niveau d’amélioration thérapeutique d’un produit médicamenteux. Dans l’ébauche des Lignes directrices révisées publiées en août 2008 aux fins de la consultation, le CEPMB a proposé quatre nouveaux niveaux d’amélioration thérapeutiques (avec l’ajout du niveau « une amélioration modeste ») et la révision des facteurs primaires et secondaires utilisés pour déterminer les niveaux d’amélioration thérapeutique.
Points de vue des intervenants
Dans les commentaires qu’ils ont formulés suite à l’Avis et commentaires du mois d’août 2008, les intervenants se sont dits favorables à la création du niveau d’amélioration « modeste » des bienfaits thérapeutiques et ont affirmé qu’il s’agissait d’une amélioration par rapport aux catégories existantes. Les commentaires des intervenants étaient toutefois partagés concernant les facteurs primaires et les facteurs secondaires, certains approuvant l’inclusion de nouveaux facteurs sur l’incidence économique alors que d’autres privilégiaient les facteurs proposés par le Conseil.
Position du Conseil
Le Conseil a décidé d’adopter les modifications proposées aux niveaux d’amélioration thérapeutique présentés dans le document d’Avis et commentaires d’août 2008. Le Conseil a également décidé d’adopter les modifications proposées concernant les facteurs primaires et secondaires de détermination des niveaux d’amélioration thérapeutique, à l’exception que les facteurs secondaires ne peuvent être appliqués que jusqu’à concurrence du niveau d’amélioration « modeste » des bienfaits thérapeutiques. Le Conseil est d’avis que l’importance relative des facteurs secondaires n’est pas suffisante pour faire passer le niveau d’amélioration thérapeutique de « modeste » à « important ». Seul l’examen des facteurs primaires peut mener à un tel changement.
Tests appliqués au prix de lancement
L’élaboration de nouveaux niveaux d’amélioration thérapeutique a obligé le CEPMB à reconsidérer les tests de prix associés. En 2008, un Groupe de travail sur les tests appliqués au prix a été constitué pour déterminer à la lumière des nouveaux niveaux d’amélioration thérapeutique proposés quelles révisions il y aurait lieu d’apporter aux tests appliqués au prix de lancement.
i) Test de la relation raisonnable
Dans l’ébauche de ses Lignes directrices révisées soumise à la consultation des intervenants en août 2008, le Conseil a retenu nombre des recommandations du Groupe de travail, dont le maintien du test de la relation raisonnable pour les produits médicamenteux constituant une extension de la gamme de produits (lorsque le breveté n’allègue aucune amélioration des bienfaits thérapeutiques ou, s’il le fait, lorsque le Groupe consultatif sur les médicaments pour usage humain ne retient pas l’allégation dans sa recommandation).
Points de vue des intervenants
Les intervenants de l’industrie ont réagi à la modification apportée au troisième volet du test de la relation raisonnable que le Conseil a présentée dans le document d’Avis et commentaires d’août 2008. Le Conseil a proposé que le prix d’un nouveau produit médicamenteux d’une concentration moindre soit proportionnel au prix du même produit d’une concentration plus grande déjà offert sur le marché canadien plutôt que de permettre un même prix à l’unité dans les deux cas. Les intervenants de l’industrie ont fait valoir que telle modification est susceptible d’amener les fabricants à ne plus offrir sur le marché des produits médicamenteux utilisés dans l’adaptation de la posologie.
Position du Conseil
Le Conseil estime que le test original de la relation raisonnable n’a pas constitué un facteur dissuasif en ce qui concerne l’adaptation posologique alors que le test révisé pourrait le faire. En conséquence, le Conseil propose de revenir à l’objet premier et à la méthodologie originale de ce test.
Suite à l’examen des différents aspects du test de la relation raisonnable, le Conseil est arrivé à la conclusion que des modifications doivent être apportées à son deuxième volet. Ces modifications visent un double objectif. Premièrement, les changements proposés reconnaissent la possibilité d’établir un même prix à l’unité posologique d’un produit médicamenteux breveté offert en différentes concentrations. Deuxièmement, les modifications proposées règlent la question d’un prix négatif possible lorsque l’intersection avec l’axe des ordonnées est négative. Le Conseil soumet ce qui suit à l’Avis et commentaires :
- Le deuxième volet du test de la relation raisonnable a été modifié pour permettre une pente nulle dans le calcul de la relation raisonnable. Cette modification a été apportée pour corriger le problème que posent les Lignes directrices existantes en vertu desquelles les produits médicamenteux affichant une relation raisonnable claire au deuxième volet du test (par exemple l’uniformisation du prix à l’unité lorsque le produit médicamenteux est offert dans différentes concentrations) est soumis au troisième volet en raison de la relation linéaire à pente nulle.
- Il est possible que la ligne représentant la relation linéaire positive la plus élevée entre les prix des concentrations existantes donne une intersection négative sur l’axe des ordonnées, ce qui sous-tend alors que le prix autorisé de certaines nouvelles concentrations se situerait sous zéro (0). Pour de tels cas, le Conseil propose de remplacer par le chiffre zéro (0) l’intersection négative sur l’axe des ordonnées et d’établir la relation linéaire en traçant une ligne entre le point d’origine (0) et le prix le plus élevé à l’unité du produit médicamenteux de comparaison.
Le Conseil a aussi élaboré davantage la description du test de la relation raisonnable afin que les intervenants puissent mieux comprendre quand et comment ce test sera appliqué.
| Sommaire des principaux changements soumis à la présente consultation |
Emplacement dans le Compendium révisé |
| Éclaircissement des circonstances dans lesquelles le test de la relation raisonnable sera appliqué et de la façon dont il le sera. |
Appendice 4 |
| Retour à l’objet premier et à la méthodologie originale du troisième volet du test de la relation raisonnable. |
Appendice 4, Test 3 |
| Le deuxième volet du test de la relation raisonnable a été modifié pour admettre la possibilité d’un même prix à l’unité pour plusieurs concentrations d’un produit médicamenteux. |
Appendice 4, Test 2, Point centré 2 |
| Le deuxième volet du test de la relation raisonnable a été modifié de façon à pouvoir remplacer l’intersection négative sur l’axe des ordonnées lorsque le point d’intersection sur cet axe se situe sous le point zéro (0). Dans ce cas, la ligne de la relation raisonnable sera retracée afin de relier le point d’origine (0) et le prix le plus élevé à l’unité du produit médicamenteux de comparaison. |
Appendice 4, Volet 2, Point centré 5 |
ii) Lignes directrices modifiées pour certains produits médicamenteux génériques brevetés
A) Comparaison selon la catégorie thérapeutique limitée
Dans son document d’Avis et commentaires d’août 2008, le Conseil a soumis à la consultation de ses intervenants une modification qui limiterait la sélection des médicaments utilisés dans la comparaison selon la catégorie thérapeutique au produit médicamenteux de référence (bioéquivalent) ou aux produits médicamenteux de marque fabriqués par la société ayant octroyé la licence. Cette proposition qui a été défendue par les représentants de l’industrie du médicament générique est perçue par le Conseil comme une approche raisonnable.
Points de vue des intervenants
Les intervenants de l’industrie du médicament générique se sont montrés favorables aux propositions formulées dans l’ébauche des Lignes directrices révisées d’août 2008. Les autres intervenants ne se sont pas vraiment opposés au fait de limiter la comparaison selon la catégorie thérapeutique au produit médicamenteux de marque de référence/fabriqué par la société ayant octroyé la licence.
Position du Conseil
Le Conseil souhaite simplifier la procédure de comparaison selon la catégorie thérapeutique pour faire en sorte qu’un produit médicamenteux générique « bioéquivalent » ou fabriqué « sous licence » ait pour unique produit de comparaison le produit médicamenteux de marque de référence ou produit de marque fabriqué par la société ayant octroyé la licence. De fait, cette limite sous-tend que les produits doivent être comparés avec la même entité chimique, la même indication ou la même utilisation, la même forme posologique ou une forme comparable et le même régime posologique. En conséquence, il y a lieu de préciser cette limite dans le texte de la section sur la relation raisonnable (chapitre 2, alinéa 2.11(c)) et dans l’appendice 4 de l’ébauche des Lignes directrices révisées. Cette proposition qui a été acceptée par les parties assujetties à la réglementation aurait pour avantage de requérir moins de temps de travail pour le personnel du Conseil et moins de ressources du Groupe consultatif sur les médicaments pour usage humain. Ce changement sera donc retenu.
ii) Lignes directrices modifiées pour certains produits médicamenteux génériques brevetés
B) Comparaison du prix au Canada avec le prix international le plus élevé
En 2008, les consultations auprès de l’industrie du médicament générique ont soulevé différentes questions apparemment particulières à la dynamique du marché et à la concurrence qui se pose aux produits médicamenteux génériques. Dans l’ébauche des Lignes directrices révisées publiées en août 2008, le Conseil a soumis à la consultation de ses intervenants une proposition selon laquelle, dans le cas d’un produit médicamenteux générique breveté bioéquivalent, le résultat de la Comparaison selon la catégorie thérapeutique primerait même si le prix au Canada du nouveau produit médicamenteux générique est plus élevé que ses prix dans les pays de comparaison. Autrement dit, le produit générique serait soustrait de la comparaison du prix au Canada avec le prix international le plus élevé.
Points de vue des intervenants
Les intervenants de l’industrie du médicament générique se sont portés à l’appui de cette proposition formulée dans l’ébauche révisée des Lignes directrices publiée en août 2008. Presque tous les autres intervenants qui ont exprimé leur point de vue sur cette question ont indiqué qu’il ne devrait pas exister une catégorie spéciale de brevetés selon le type de société qui vend le produit (produits génériques versus produits de marque/innovateurs) et que les brevetés devaient tous être traités sur un même pied d’égalité. Ils ont enjoint le Conseil de ne pas permettre telle exemption à la comparaison du prix au Canada avec le prix international le plus élevé.
Position du Conseil
Le Conseil a rencontré les représentants de l’industrie du médicament générique et les a invités à soumettre leurs représentations sur ce point. Le Conseil convient que l’équité est un principe prépondérant du processus de réglementation des prix et, en conséquence, tous les brevetés devraient être assujettis à la contrainte que pose la comparaison du prix au Canada avec le prix international le plus élevé.
| Sommaire des principaux changements soumis à la consultation |
Emplacement dans le Compendium révisé |
| Retirer de l’ébauche du Compendium révisé l’exception prévoyant que les produits médicamenteux génériques « bioéquivalents » pourraient être soustraits de la comparaison du prix au Canada avec le prix international le plus élevé. |
Retiré de : Partie III, Chapitre 2, paragraphe 2.11, alinéa C, point centré 1 |
iii) Comparaison du prix selon la catégorie thérapeutique internationale
En 2007, le Conseil a chargé le Groupe de travail sur la comparaison selon la catégorie thérapeutique internationale de formuler des recommandations concernant la sélection des médicaments pour la comparaison du prix selon la catégorie thérapeutique internationale. Après avoir pris connaissance des recommandations du Groupe de travail, le CEPMB a proposé dans son document d’Avis et commentaires d’août 2008 que la comparaison du prix selon la catégorie thérapeutique internationale ne soit pas un test principal, mais qu’il serve plutôt à fournir des éléments d’information dans le contexte d’une enquête sur le prix d’un produit médicamenteux qui semble excessif. Le Conseil a également proposé d’utiliser les deux variantes suivantes pour la comparaison, à savoir 1) « l’approche du ratio » et 2) « l’approche classique de la catégorie thérapeutique ».
Points de vue des intervenants
Les intervenants du secteur de l’industrie ont exprimé certaines réticences du fait que le CEPMB n’avait pas adopté la recommandation du Groupe de travail sur la comparaison du prix selon la catégorie thérapeutique internationale qui prévoyait que les produits médicamenteux génériques ne seraient pas pris en compte dans la comparaison du prix selon la catégorie thérapeutique internationale lorsque le Conseil estime que le résultat pertinent est établi au moyen d’une mesure se situant sous le prix le plus élevé obtenu avec la comparaison du prix selon la catégorie thérapeutique internationale.
Position du Conseil
Le Conseil a réévalué ce test et propose les changements suivants :
- Lorsque l’approche de la « catégorie classique » et du « ratio » est utilisée, le CEPMB propose d’utiliser le prix international médian ainsi obtenu comme mesure du prix excessif.
- Lorsque la comparaison du prix selon la catégorie thérapeutique internationale est appliquée, les produits médicamenteux génériques utilisés pour la comparaison du prix selon la catégorie thérapeutique internationale se limiteront aux produits vendus par les sociétés qui vendent également le même produit médicamenteux générique au Canada.
La première modification vise à communiquer très clairement aux intervenants la mesure qui sera prise pour la comparaison du prix selon la catégorie thérapeutique internationale. De l’avis du Conseil, l’élaboration d’une « série de valeurs statistiques » comme il a déjà été fait mention ne constitue pas une avenue très utile. Les intervenants auront avantage à connaître à l’avance la mesure statistique prédominante dans la comparaison du prix selon la catégorie thérapeutique internationale.
Quant à la deuxième modification, elle répond aux préoccupations exprimées par l’industrie concernant l’inclusion des produits médicamenteux génériques dans les comparaisons du prix selon la catégorie thérapeutique internationale. Le Conseil ne privilégie pas l’exclusion totale des produits médicamenteux génériques de la comparaison du prix selon la catégorie thérapeutique internationale, mais considérant le nombre important de produits médicamenteux génériques dans les pays de comparaison, le Conseil se doit de reconnaître que l’inclusion de tous les produits médicamenteux génériques pourrait fort bien fausser les résultats de la comparaison. Afin de s’assurer que les médicaments utilisés pour la comparaison sont pertinents et appropriés, le prix du produit médicamenteux sous examen ne sera comparé qu’aux prix des produits médicamenteux génériques qui sont vendus par la même société au Canada et dans les pays de comparaison.
| Sommaire des principales modifications soumises à la consultation |
Emplacement dans le Compendium révisé |
| Lorsque l’approche de la « catégorie classique » et l’approche du « ratio »sont toutes deux utilisées dans la comparaison du prix selon la catégorie thérapeutique internationale, le prix international médian obtenu sera comparé au prix de transaction moyen national du produit médicamenteux du breveté. |
Appendice 7, Section 3 |
| La sélection des produits médicamenteux génériques utilisés pour la comparaison du prix selon la catégorie thérapeutique internationale sera limitée aux produits qui sont vendus par la même société au Canada et dans les pays de comparaison. |
Appendice 7, Section 2 |
Examens du prix sur un marché
Dans le cours des consultations précédentes, certains intervenants ont exprimé l’avis que la capacité de certaines catégories de clients ou de certaines provinces de négocier des prix avantageux pour leurs produits médicamenteux pouvait être liée au fait que d’autres clients/provinces paient leurs produits médicamenteux à des prix qui pourraient être considérés excessifs. Pour répondre à cette préoccupation, le Conseil a formulé dans le document d’Avis et commentaires d’août 2008 des propositions concernant l’examen du prix sur « un marché », qui consistaient nommément à veiller à ce que les prix de lancement sur des marchés donnés ne soient pas excessifs et, dans le cas des produits médicamenteux existants, à effectuer des examens de prix au cas par cas au niveau du marché lorsque la variabilité des prix des différents marchés semblent causer un problème. Le Conseil a également sollicité les points de vue des intervenants sur la méthodologie qui conviendrait le mieux pour le calcul des recettes excessives lorsque le prix du produit est excessif sur un marché donné.
Points de vue des intervenants
Les intervenants ont exprimé des points de vue partagés en ce qui concerne l’examen du prix au niveau du marché. D’une façon générale, les intervenants de l’industrie étaient d’avis que l’utilité d’une telle analyse n’avait pas été suffisamment démontrée alors que les autres intervenants opinaient que tels examens de prix étaient nécessaires. Toutefois, il est ressorti des nombreux mémoires reçus que le texte décrivant les propositions concernant l’examen du prix sur un marché était vague et nébuleux.
Position du Conseil
En principe, le Conseil est d’avis que l’examen du prix d’un marché s’inscrit dans son mandat. Il a été clairement démontré dans le Guide de discussion aux fins de la consultation publié en mai 2006 que certains marchés paient des prix de lancement dépassant de 25 % et plus le Prix moyen maximal potentiel (auparavant appelé Prix maximum non excessif pour la période de lancement).
Le Conseil reconnaît qu’il y a lieu de clarifier l’approche des examens du prix sur un marché. La toute dernière version de l’ébauche révisée des Lignes directrices expose d’une façon plus claire et plus détaillée les examens de prix sur un marché. D’ailleurs, ce sujet fait l’objet d’un nouvel appendice qui décrit la méthodologie.
En plus de faire l’examen du prix national et des prix pour les trois catégories de clients, le Conseil propose de faire l’examen du prix de lancement au niveau de chaque province et de chaque territoire.
En ce qui concerne les produits médicamenteux existants, le Conseil continuera de faire l’examen du prix au niveau national, mais il n’effectuera des examens du prix au niveau de la catégorie de clients et (ou) de la province/territoire que si le Prix de transaction moyen national semble excessif ou s’il reçoit une plainte.
En ce qui concerne la méthodologie de calcul des recettes excessives, les intervenants ont indiqué que ces recettes devraient être calculées à l’aide du prix moyen du produit sur tous les marchés au Canada (Prix de transaction moyen national) de manière à reconnaître et à prendre en compte les prix moins élevés pratiqués sur d’autres marchés plutôt que de limiter le calcul au marché où les prix pratiqués étaient excessifs. Le Conseil souscrit à cette position et a modifié en conséquence le texte de l’ébauche révisée de ses Lignes directrices.
| Sommaire des principaux changements soumis à la consultation |
Emplacement dans le Compendium révisé |
| Un nouvel appendice (l’appendice 12) a été ajouté au document afin de clarifier l’approche de l’examen du prix sur un marché. |
Appendice 12 |
| Augmentation du nombre de marchés qui, pour la période de lancement, feront l’objet d’un examen du prix sur un marché. Ces marchés supplémentaires sont les suivants : chaque province et chaque territoire en plus des trois catégories de clients (hôpital, pharmacie et grossiste) et le niveau national. |
Partie III, Chapitre 2, alinéa 2.13 |
| Les recettes excessives seront calculées sur la base du prix national pour tous les marchés au Canada (Prix de transaction moyen national). |
Appendice 12, Point centré 7, Sous-point centré 2 |
Révision du Prix moyen non excessif après le lancement du produit médicamenteux sur le marché
Dans le document de discussion publié en janvier 2008, le CEPMB a fait état de trois circonstances où il pourrait être approprié de « réviser » un Prix moyen non excessif, à savoir : 1) lorsque sont présentées de nouvelles données scientifiques; 2) lorsque le nouveau produit médicamenteux était vendu dans un nombre trop restreint de pays de comparaison et que le test du prix international médian constituait le test principal appliqué au prix de lancement; et 3) en raison des arguments invoqués concernant les coûts potentiels de réalisation et de mise en marché.
Dans le document d’Avis et commentaires d’août 2008, le Conseil a pris la position suivante :
- En ce qui concerne les nouvelles données scientifiques, le Groupe consultatif sur les médicaments pour usage humain serait appelé à identifier les lacunes ou les failles importantes des données scientifiques, mais il n’y avait pas lieu d’adopter une ligne directrice sur ce sujet même.
- Lorsqu’un nouveau produit médicamenteux breveté est vendu dans un nombre trop restreint de pays de comparaison et que le test du prix international médian constitue le test principal appliqué au prix de lancement, le statu quo semble l’approche appropriée (révision du prix international médian pour les trois années qui suivent la période de lancement ou au moment où le produit médicamenteux breveté est vendu dans cinq pays de comparaison, soit la première de ces deux éventualités).
- L’examen du prix en relation avec les coûts de réalisation et de mise en marché peut être approprié, mais il ne s’agit pas d’un facteur de prix dont le Conseil doit obligatoirement prendre en compte (en vertu de l’alinéa 85(1) de la Loi). Une certaine latitude de la part du personnel du Conseil a été permise, mais aucune ligne directrice particulière n’a été proposée.
Points de vue des intervenants
Il ressort des mémoires reçus suite au document d’Avis et commentaires d’août 2008 que les intervenants approuvent en général l’orientation du Conseil. Les intervenants ont toutefois noté l’absence dans l’ébauche des Lignes directrices révisées d’août 2008 de la clause citée ci-après qui figure dans les Lignes directrices existantes :
« Le CEPMB reconnaît que le prix de référence d’un produit médicamenteux vendu à titre de drogue nouvelle de recherche ou au titre du Programme d’accès spécial doit être révisé après la réception de son Avis de conformité. Dans un tel cas, le prix de transaction moyen est vérifié afin de voir s’il semble excessif aux termes des Lignes directrices qui s’appliquent aux nouveaux produits médicamenteux. »
Position du Conseil
Le Conseil saisit bien les préoccupations des intervenants qui souhaiteraient que le Prix moyen non excessif d’un produit médicamenteux soit révisé après réception de son Avis de conformité, mais il estime que l’obtention de l’Avis de conformité ne justifie pas à elle seule une révision du Prix moyen non excessif. Le Conseil reconnaît qu’il peut être pertinent de réviser le Prix moyen non excessif d’un produit médicamenteux sur la base de son coût de réalisation et de mise en marché. De tels arguments pourraient être invoqués lorsque le produit a été vendu sur le marché canadien pour des raisons humanitaires, à titre de nouvelle drogue de recherche ou par le truchement du Programme spécial d’accès et, ayant obtenu son Avis de conformité, le breveté a dû encourir des coûts pour lancer son produit sur le marché canadien. Le texte sur ce point a été intégré dans l’ébauche révisée des Lignes directrices aux fins d’un Avis et commentaires.
| Sommaire des principales modifications soumises à la consultation |
Emplacement dans le Compendium révisé |
| Ajout du paragraphe suivant dans les Lignes directrices : « Le Conseil est conscient que le breveté peut invoquer les coûts de réalisation et de mise en marché pour justifier un rajustement du prix moyen non excessif de son produit médicamenteux breveté (c.-à-d. lorsque l’Avis de conformité a été attribué au produit médicamenteux et que ce produit a été initialement vendu pour des motifs humanitaires à titre de nouvelle drogue de recherche, par le truchement d’une demande d’essai clinique ou, encore, au titre du Programme spécial d’accès).» |
Partie III, Chapitre 2, Paragraphe 3.5 |
Prise en compte des avantages (Méthodologie de la majoration)
Dans le document d’Avis et de commentaires d’août 2008, le Conseil a clairement mentionné qu’il ne cherchait aucunement à dissuader les brevetés d’offrir des avantages à leurs clients et a proposé comme solution de rechange à la méthodologie du prix rajusté selon l’IPC une nouvelle méthodologie qu’on a appelée « Méthodologie de la majoration ». Cette nouvelle méthodologie se fonde sur le principe que lorsque le prix semble excessif seulement en raison de la cessation ou de la réduction d’un avantage et que le breveté fournit des éléments de preuve à l’appui, le Prix de transaction moyen obtenu ne devrait alors pas être considéré excessif si le prix ne fait que revenir à son prix d’avant l’avantage.
Points de vue des intervenants
Les intervenants de l’industrie ont exprimé de nombreuses réserves en ce qui concerne la méthodologie de la majoration, dont les suivantes : la méthodologie est beaucoup trop complexe, la méthodologie fera augmenter la charge de travail pour les brevetés et pour les membres du personnel, il n’est pas aussi facile que le prétend la méthodologie de faire un suivi minutieux des avantages offerts aux clients. Il a également été recommandé de constituer un comité formé de représentants du CEPMB et de Rx&D pour discuter des défis que la proposition pourrait poser pour l’industrie. Le comité spécial constitué de représentants de Rx&D et du CEPMB s’est réuni à trois occasions pour discuter des différents sujets de préoccupation de l’industrie.
Position du Conseil
La position du Conseil est qu’il y a lieu de se doter d’une méthodologie qui minimise l’effet dissuasif qu’est susceptible de causer l’obligation imposée aux brevetés de comptabiliser dans leurs rapports semestriels les avantages qu’ils ont consentis à leurs clients. Essentiellement, la méthodologie de la majoration accorde une certaine latitude aux brevetés dont les prix, nets de la valeur des avantages qu’ils ont consentis à leurs clients, auraient dû être soumis à la méthodologie du prix rajusté selon l’IPC après avoir cessé d’offrir tels avantages. La nouvelle ébauche révisée des Lignes directrices jette plus de lumière sur la méthodologie de la majoration :
- Descriptions plus claires de la mécanique de la nouvelle méthodologie
- Plus grande souplesse pour permettre au Prix de transaction moyen d’un marché de « rebondir » au prix auquel il était vendu avant l’avantage sur le même marché si, bien sûr, le breveté fournit les éléments de preuve requis
- Plus grande souplesse pour permettre au prix d’un marché qui a reçu un avantage, à compter de la date de la première vente et, par la suite, pour chaque période de rapport, de « rebondir » au prix non excessif sur un autre marché qui n’a jamais reçu l’avantage si, bien sûr, le breveté fournit les éléments de preuve requis, et
- Lignes directrices concernant les attentes minimales des brevetés en termes d’éléments de preuve que les brevetés devront fournir pour que leur prix puisse être soumis à la méthodologie de la majoration.
| Sommaire des principales modifications soumises à la consultation |
Emplacement dans le Compendium révisé |
| Un nouvel appendice a été ajouté afin de fournir des éclaircissements sur la méthodologie de la majoration. |
Appendice 10 |
Utilisation des produits médicamenteux brevetés et non brevetésdans les tests appliqués au prix
Dans le cours du processus d’examen du prix d’un nouveau produit médicamenteux, le personnel du Conseil fait également l’examen des produits médicamenteux de comparaison clés, brevetés ou non brevetés, afin de vérifier si ces prix sont excessifs aux termes des Lignes directrices sur les prix excessifs. Même si le personnel du Conseil applique cette procédure depuis déjà un certain temps, il n’existe encore aucun énoncé de politique qui officialise la procédure.
Position du Conseil
Le Conseil a intégré dans le document de consultation une politique sur l’utilisation des produits médicamenteux brevetés et non brevetés dans les tests appliqués au prix et ce, afin de mieux informer les intervenants sur les pratiques courantes du personnel du Conseil. Le Conseil a également ajouté dans les Lignes directrices de plus amples détails concernant la façon dont sera fait l’examen des prix des produits médicamenteux brevetés et non brevetés aux fins de déterminer s’ils semblent ou non excessifs.
| Sommaire des principales modifications soumises à la consultation |
Emplacement dans le Compendium révisé |
| Ajout dans le Compendium d’une politique sur l’utilisation des produits médicamenteux brevetés et non brevetés dans les tests appliqués au prix |
Partie II, Section 6 |
| Ajout de plus amples détails concernant la façon dont sera fait l’examen des prix des produits médicamenteux brevetés et non brevetés aux fins de déterminer s’ils semblent ou non excessifs. |
Appendice 3, Section 2, Paragraphes 3-5
Appendice 4, Paragraphes 5-8 |
Remboursement des recettes excessives
En vertu des Lignes directrices existantes, le breveté tire des recettes excessives de la vente de son produit lorsque le Prix de transaction moyen de son produit médicamenteux est plus élevé que son Prix maximum non excessif. Même si les Lignes directrices actuelles traitent de la façon dont les recettes excessives peuvent être remboursées, le Conseil saisit l’occasion que lui fournit le présent exercice de révision des Lignes directrices pour faire un peu plus de lumière sur les avenues qui s’offrent aux brevetés tenus de rembourser des recettes excessives.
Position du Conseil
Dans le cadre de la présente consultation, le Conseil propose un énoncé de politique sur le remboursement des recettes excessives ainsi qu’un appendice consolidé. Les intervenants devraient prendre bonne note des modifications suivantes que le Conseil a proposées :
- Pour rembourser les recettes excessives au moyen d’une réduction du prix du produit médicamenteux, le prix moyen du produit médicamenteux ne sera réputé avoir été réduit que s’il est moins élevé que le Prix moyen non excessif de l’année précédente. Le breveté ne sera pas autorisé à renoncer à l’augmentation du prix auquel il a droit comme mesure de remboursement des recettes excessives qu’il a tirées de la vente de son produit à un prix excessif.
La logique de cette modification est que les recettes excessives encaissées par le breveté démontrent que les clients en général ont acheté le produit médicamenteux à un prix qui semblait excessif et que, en application de l’article 83 de la Loi, le breveté doit réduire le prix de son produit afin de rembourser les recettes excessives.
- Les recettes excessives dont la valeur ne justifie pas la tenue d’une enquête qui sont encaissées pendant six semestres de rapport consécutifs (3 ans) devront être remboursées en vertu d’un Engagement de conformité volontaire. À défaut du breveté de soumettre tel engagement au Conseil, le personnel du Conseil portera l’affaire à l’attention du président.
La logique de cette modification est que, en vertu des Lignes directrices existantes, les brevetés dont la valeur des recettes excessives qu’ils ont encaissées ne justifie pas la tenue d’une enquête peuvent conserver ces recettes indéfiniment. Selon une recherche menée à l’interne par le personnel du Conseil, il n’est pas inhabituel que telles recettes excessives soient encaissées pendant plusieurs années.
- L’exception suivante à la méthodologie du prix rajusté selon l’IPC s’applique lorsque la réduction du prix sous le Prix moyen non excessif est appliquée sur un ou sur plusieurs marchés aux fins de rembourser les recettes excessives encaissées. Après que le breveté aura remboursé en totalité les recettes excessives qu’il a encaissées, les Prix de transaction moyens du produit médicamenteux sur ces marchés pourront dès la prochaine période de rapport revenir aux Prix moyens non excessifs du marché auxquels ils étaient avant que soit appliquée la réduction du prix.
La logique de cette modification est que le Conseil ne souhaite pas créer une situation où les brevetés seraient dissuadés de rembourser les recettes excessives au moyen d’une réduction du prix de leur produit.
| Sommaire des principales modifications soumises à la consultation |
Emplacement dans le Compendium révisé |
| Nouvelle politique sur le remboursement des recettes excessives |
Partie II, Section 7 |
| Nouvel appendice soumis aux commentaires des intervenants sur la politique proposée concernant le remboursement des recettes excessives |
Appendice 13 |
Préambule
Le Conseil d’examen du prix des médicaments brevetés (CEPMB) souhaite rendre son processus d’examen du prix des médicaments encore plus ouvert et plus transparent afin de continuer à bien servir les meilleurs intérêts de ses intervenants.
Le Compendium des Politiques, des Lignes directrices et des Procédures (le Compendium) a pour objectif principal d’informer les brevetés sur ses politiques, ses lignes directrices et ses procédures qui régissent les examens des prix des produits médicamenteux brevetés vendus au Canada ainsi que sur ses procédures d’examen scientifique et d’examen des prix qui semblent excessifs.
Le CEPMB estime parfois nécessaire de mettre à jour ses Lignes directrices afin qu’elles demeurent et qu’elles facilitent l’application de ses principes d’équité, de transparence, d’ouverture et de prévisibilité. Dans un tel cas, le CEPMB consulte ses intervenants au moyen de son processus d’Avis et de commentaires.
Le Compendium est organisé comme suit :
Partie I – Le cadre juridique
Partie II – Les politiques
Partie II – Les Lignes directrices et procédures
Chapitre 1 – Le processus d’examen scientifique
Chapitre 2 – Le processus d’examen du prix
Chapitre 3 – Les enquêtes
Appendices
Partie I – Le cadre juridique
1. Origine du CEPMB
1.1 Le CEPMB a été créé en application des modifications apportées à la Loi sur les brevets (la « Loi ») entrées en vigueur le 7 décembre 1987. Avant 1987, le Canada a cherché à réduire les prix des médicaments brevetés en favorisant la concurrence au moyen de licences obligatoires. Les modifications de 1987 ont amélioré la protection qu’un brevet confère aux médicaments afin que les brevetés investissent davantage dans la recherche-développement (R-D) au Canada. C’est d’ailleurs de ces modifications qu’est né le CEPMB en vertu de la politique de protection du consommateur. Le rôle du CEPMB est de s’assurer que les médicaments brevetés ne sont pas vendus au Canada à des prix excessifs.
1.2 D’autres modifications à la Loi, celles-là en vigueur depuis le 15 février 1993, ont aboli le régime de licences obligatoires pour les médicaments brevetés, accordant ainsi aux brevetés un monopole sur le marché canadien pendant toute la durée du brevet. Pour combler le vide créé par l’abolition des licences obligatoires, les modifications de 1993 ont aussi élargi les pouvoirs correcteurs du CEPMB. Depuis, le Conseil peut, lorsque les circonstances l’exigent, obliger les brevetés à rembourser les recettes excessives qu’ils ont tirées de la vente de leur produit médicamenteux à un prix excessif et ce, en plus de baisser le prix de leur médicament.
2. Mandat du CEPMB
2.1 Le mandat du CEPMB comporte les deux volets suivants :
- Réglementation – Veiller à ce que les prix auxquels les brevetés rendent leurs médicaments brevetés vendus au Canada ne soient pas excessifs, conformément aux intérêts des consommateurs et du régime canadien de soins de santé.
- Rapport – Chaque année, faire rapport au Parlement des activités du CEPMB, des ratios des dépenses de R-D des brevetés et de l’industrie des médicaments brevetés par rapport aux recettes tirées des ventes, des tendances des prix de tous les médicaments afin d’éclairer les processus de prise de décisions et d’élaboration de politiques du régime de soins de santé. En vertu de l’article 90 de la Loi, préparer des analyses et des rapports sur d’autres sujets que lui défère le ministre de la Santé.
3. Structure et fonctionnement du CEPMB
3.1 Le CEPMB est un organisme indépendant et autonome qui détient des pouvoirs quasi judiciaires. Afin de garantir l’indépendance et l’autonomie du Conseil, la Loi ne confère au gouvernement aucun pouvoir explicite ou implicite qui lui permettrait d’avoir un droit de regard sur les activités du Conseil ou, encore, de réviser ses décisions et ses ordonnances. De même, le CEPMB ne participe d’aucune façon au processus d’élaboration des politiques.
3.2 Comme le prévoient les principes du droit administratif, les décisions de fond rendues par le Conseil peuvent être soumises à la révision judiciaire de la Cour fédérale du Canada sur toute question de fond ou d’ordre procédural.
3.3 Le CEPMB est régi par un Conseil formé de cinq membres nommés en vertu du paragraphe 91(1) de la Loi. Par ailleurs, les membres du personnel sont nommés en vertu du paragraphe 94(1) de la Loi.
3.4 Le CEPMB est habilité à se doter de politiques et de procédures qui régissent la façon dont il s’acquitte de ses obligations réglementaires. À cette fin, pour lui permettre de s’acquitter de ses obligations d’une façon juste et équitable, le CEPMB a opté pour une séparation administrative des fonctions d’examen et des fonctions de poursuite. Les fonctions d’examen et de poursuite sont menées par le personnel du Conseil tandis que la compétence des membres du Conseil est de décider d’une affaire portée à leur attention.
3.5 Le personnel du Conseil vaque aux activités courantes du CEPMB, dont l’administration du Règlement sur les médicaments brevetés (le Règlement) en s’assurant que les brevetés soumettent dans les délais impartis les rapports exigés. Il effectue également l’examen des prix des médicaments brevetés de la façon prescrite dans les Lignes directrices approuvées par le Conseil.
3.6 S’il arrive à la conclusion qu’il est dans l’intérêt public de tenir une audience en vertu du paragraphe 83(6) de la Loi aux fins de décider si un médicament breveté est ou a été vendu sur un marché au Canada à un prix réputé excessif, le président du Conseil émet un Avis d’audience et forme un panel d’audience constitué de membres du Conseil.
3.7 Afin d’assurer l’impartialité du panel d’audience, aucun membre du Conseil autre que le président n’est informé des résultats des vérifications de prix effectuées par le personnel du Conseil avant que l’affaire ne soit entendue dans le cadre d’une audience publique. Seul le président est informé de ces résultats en sa capacité de chef de la direction du CEPMB puisqu’il lui appartient de décider, en application du paragraphe 93(2) de la Loi, s’il est dans l’intérêt public de tenir une audience.
4. Compétence du CEPMB en matière de réglementation des prix
4.1 La Loi investit le CEPMB du pouvoir de déterminer si le breveté ou l’ancien breveté d’une invention liée à un médicament, vend ou a vendu sur un marché au Canada son médicament à un prix jugé excessif et ce, lorsque les critères suivants s’appliquent1 :
4.1.1 Breveté ou ancien breveté
- Aux termes du paragraphe 79(1) de la Loi, le mot « breveté » désigne la personne ayant pour le moment droit aux retombées d’un brevet pour une invention liée à un médicament, ainsi que quiconque était titulaire d’un brevet pour telle invention ou exerce ou a exercé les droits d’un titulaire dans un cadre autre qu’une licence accordée avant le 20 décembre 1991 par le Commissaire aux brevets et qui n’était pas arrivée à échéance le 15 février 1993, date de l’entrée en vigueur de la Loi de 1992 modifiant la loi sur les brevets.
- Le CEPMB a également compétence sur l’ancien breveté d’une invention pour la période durant laquelle il était détenteur du brevet.
4.1.2 Brevet lié au médicament
Médicament
- Le mot « médicament » n’est pas définit dans la Loi sur les brevets. Veuillez consulter le politique du Conseil à l’égard du sens donné au mot.
Brevet
- Le paragraphe 79(2) de la Loi prévoit qu’un brevet pour une invention est lié à un médicament si l’invention est destinée à des médicaments ou à la préparation ou à la production de médicaments ou est susceptible d’être utilisée à de telles fins.
- Le CEPMB désigne par le mot « brevet » tout brevet canadien pour une invention liée à un médicament. Sans être limitatif, cette définition couvre les brevets suivants :
- Brevets pour des ingrédients actifs
- Brevets pour des procédés de fabrication
- Brevets pour un procédé d’administration ou pour une forme posologique faisant partie intégrante du procédé d’administration du médicament
- Brevets pour des indications/utilisations, et
- Brevets pour des formulations médicinales.
- Un brevet est lié à un médicament s’il peut être utilisé pour la préparation ou la production de médicaments, qu’il le soit ou non.
- À priori, il doit exister un rapport ou un lien logique même tenu2 entre l’invention décrite dans le brevet et le médicament.
4.1.3 Vente sur un marché au Canada
- Le breveté ou l’ancien breveté doit vendre ou avoir vendu son médicament breveté sur un marché au Canada.
- Exception faite des médicaments vendus en vertu d’une licence obligatoire accordée par le Commissaire aux brevets avant le 20 décembre 1991 qui n’était pas arrivée à échéance en date du 15 février 1993, date d’entrée en vigueur de la Loi de 1992 modifiant la Loi sur les brevets, tous les médicaments brevetés vendu sur un marché au Canada pour usage humain ou pour usage vétérinaire sont assujettis à la compétence du CEPMB en matière d’examen du prix, incluant les médicaments brevetés ayant reçu leur Avis de conformité ou vendus en vertu du Programme spécial d’accès, faisant l’objet d’une Demande d’essai clinique ou, encore, comme Drogue nouvelle de recherche.
- Le CEPMB fait l’examen du prix du premier médicament que le breveté a vendu directement à une catégorie de clients (grossiste, hôpital, pharmacie ou autres) au Canada. Le CEPMB n’a pas droit de regard sur les prix auxquels les grossistes ou les détaillants vendent les médicaments brevetés ni sur les honoraires que chargent les pharmaciens.
- Le breveté n’est pas tenu de faire approuver par le CEPMB le prix de vente au Canada de son médicament breveté avant de l’offrir sur le marché canadien. Toutefois, le breveté peut demander au personnel du Conseil de vérifier le prix de son médicament avant de le lancer sur le marché canadien afin de s’assurer qu’il n’est pas excessif.
- Le CEPMB ne fixe pas les prix de vente au Canada des médicaments brevetés. Toutefois, il appartient au Conseil de déterminer le prix moyen maximal auquel le breveté peut vendre son produit (prix moyen maximal possible) au Canada ainsi que le prix moyen considéré non excessif.
5. Facteurs de réglementation des prix
5.1 Le paragraphe 85(1) de la Loi présente les facteurs dont le Conseil doit tenir compte dans le cadre d’une audience où il est appelé à déterminer si le breveté ou l’ancien breveté vend ou a vendu sur un marché au Canada son médicament breveté à un prix excessif. Ces facteurs sont les suivants :
- le prix de vente du médicament sur un tel marché
- le prix de vente d’autres médicaments de la même catégorie thérapeutique sur un tel marché
- le prix de vente à l’étranger du médicament et d’autres médicaments de la même catégorie thérapeutique
- les variations de l’indice du prix à la consommation
- tous les autres facteurs mentionnés dans le règlement d’application.
5.2 Si, après avoir considéré les facteurs susmentionnés, le Conseil n’est pas en mesure de déterminer si le prix de vente du médicament sous examen est ou non excessif, il peut, en vertu du paragraphe 85(2) tenir compte des coûts de réalisation et de mise en marché du médicament ainsi que d’autres facteurs précisés dans le règlement d’application ou qu’il estime pertinents.
5.3 En application du paragraphe 96 (5) de la Loi, le Conseil a consulté tous ses intervenants avant de formuler ses Lignes directrices en vertu du paragraphe 96(4). Même si le Conseil et les brevetés ne sont pas strictement assujettis à ces Lignes directrices, elles proposent néanmoins une approche et une méthode d’application des facteurs de prix énumérés dans le paragraphe 85(1) de la Loi.
6. Recours
6.1 Si le Conseil estime que le breveté vend son produit médicamenteux breveté sur un marché au Canada à un prix excessif, il peut rendre une ordonnance pour obliger le breveté à réduire le prix maximal auquel il vend son produit sur ce marché.
6.2 De plus, si le Conseil arrive à la conclusion que le breveté ou l’ancien breveté a vendu un produit médicamenteux breveté sur un marché au Canada à un prix excessif, le Conseil peut rendre une ordonnance pour obliger le breveté à rembourser la valeur estimée des recettes excessives qu’il a tirées de la vente de son produit à un prix excessif.
6.3 Si le Conseil arrive à la conclusion que le breveté ou l’ancien breveté s’est livré à une politique de vente du médicament à un prix excessif, le Conseil peut rendre une ordonnance pour obliger le breveté ou l’ancien breveté à rembourser le double de la valeur estimée des recettes excessives qu’il a tirées de la vente de son produit médicamenteux à un prix excessif.
6.4 Aux fins du remboursement des recettes excessives, le Conseil peut obliger le breveté ou l’ancien breveté :
- à réduire le prix auquel il vend son produit médicamenteux sur un marché canadien
- à réduire le prix de vente d’un autre de ses produits médicamenteux brevetés sur un marché canadien
- à rembourser les recettes excessives qu’il a tirées de la vente de son produit médicamenteux à un prix excessif au moyen d’un paiement à Sa Majesté du chef du Canada.
7. Rapports que les brevetés doivent soumettre aux fins de la réglementation du prix
7.1 Pour arriver à bien s’acquitter de son mandat, le CEPMB doit avoir accès à des renseignements fiables et à jour sur les médicaments brevetés.
7.2 La Loi et le Règlement précisent les rapports que les brevetés et les anciens brevetés pour une invention liée à un médicament breveté assujetti à la compétence du CEPMB en matière d’examen du prix doivent soumettre au CEPMB. Vous trouverez dans le Guide du breveté de plus amples renseignements concernant les différents éléments d’information dont il faut faire rapport au Conseil ainsi que sur la façon de les soumettre et les dates auxquelles ils doivent être soumis.
Notification de l’intention de vendre un médicament breveté au Canada
- L’article 82 de la Loi prévoit que le breveté doit, dans les meilleurs délais, notifier le CEPMB de son intention de vendre un médicament breveté sur un nouveau marché canadien et de la date à laquelle il compte le faire.
- Le Conseil peut, au moyen d’une ordonnance, obliger un breveté à lui fournir des renseignements concernant le prix auquel il prévoit vendre son médicament breveté au Canada. Ces renseignements ne sont exigibles que 60 jours avant la date à laquelle le médicament devrait être lancé sur le marché canadien.
Formulaire 1 (Renseignements identifiant le médicament)
- En vertu du paragraphe 3(1) du Règlement, le breveté ou l’ancien breveté d’une invention liée à un médicament doit faire rapport au CEPMB des renseignements permettant d’identifier son médicament (formulaire 1). Le formulaire 1 doit être rempli pour tous les médicaments brevetés pour usage humain ou pour usage vétérinaire et être soumis au CEPMB en même temps que la monographie du médicament breveté faisant l’objet du rapport. Toutefois, si le médicament n’a pas encore obtenu son Avis de conformité, le breveté doit soumettre les renseignements que contiendrait normalement la monographie du médicament.
- En vertu des paragraphes 3(2) et 3(3) du Règlement, les renseignements demandés sur le formulaire 1 doivent être soumis au CEPMB si le médicament n’a pas encore obtenu son Avis de conformité ou, encore, si le médicament est offert sur le marché canadien. Ces renseignements doivent être fournis au CEPMB dans les sept jours qui suivent la date d’attribution de l’Avis de conformité du médicament ou, encore, dans les sept jours qui suivent la date de la première vente du médicament au Canada, soit la première de ces deux éventualités.
- Si le breveté ou l’ancien breveté a commencé à vendre le médicament au Canada avant d’avoir obtenu son brevet, il doit remplir le formulaire 1 et le soumettre au CEPMB dans les meilleurs délais après l’émission du brevet.
Formulaire 2 (Renseignements identifiant le médicament et renseignements sur son prix)
- En vertu du paragraphe 4(1) du Règlement, le breveté ou l’ancien breveté pour une invention liée à un médicament qui vend ou qui a vendu son médicament sur un marché au Canada doit faire rapport au CEPMB des renseignements identifiant son médicament ainsi que des renseignements sur son prix (formulaire 2). Ces renseignements doivent nommément faire état de la date de la première vente du médicament au Canada, de la quantité vendue dans sa forme posologique finale, du prix de vente moyen par emballage ou, encore, des recettes nettes tirées des ventes de chaque forme posologique, concentration et emballage du médicament et ce, pour chaque catégorie de clients de chaque province et territoire.
- En vertu du paragraphe 4(4) du Règlement, c’est le prix réel ou les recettes réelles après déduction de la taxe de vente fédérale et de la valeur des avantages consentis tels que rabais, escomptes, remboursements, biens ou services gratuits, cadeaux et tout autre avantage de semblable nature qui doit être utilisé pour le calcul du prix moyen de l’emballage ou des revenus nets.
- Le paragraphe 4(2) du Règlement prévoit que lorsque le médicament pour usage humain contient une substance contrôlée au sens qu´en donne la Loi réglementant certaines drogues et autres substances comme, par exemple, des opioïdes, des amphétamines, des barbituriques et des benzodiazépines, que le médicament figure sur la liste des substances décrites dans les appendices C ou D de la Loi sur les aliments et drogues telles que des produits radiopharmaceutiques, des vaccins, des produits sanguins et des insulines ou, encore, dans l’appendice F du Règlement sur les aliments et les drogues tels que des médicaments d’ordonnance, le breveté doit fournir les renseignements demandés sur le formulaire 2 dans les 30 jours qui suivront la date de la première vente au Canada (ventes de la première journée de vente). Par la suite, le breveté ou l’ancien breveté devra présenter les mêmes renseignements au CEPMB dans les 30 jours qui suivront la fin de chaque semestre, commençant le 1er janvier et le 1er juillet de chaque année, y compris la dernière période partielle.
- L’alinéa 4(3) du Règlement prévoit que, dans le cas des médicaments pour usage humain qui ne contiennent aucune substance contrôlée ni aucune substance nommée ou décrites dans les annexes citées dans le paragraphe 4.2, incluant les médicaments pour usage humain en vente libre ou tous les médicaments pour usage vétérinaire, le breveté doit faire rapport des renseignements demandés sur le formulaire 2 pour toutes les périodes de vente. Ces renseignements doivent être fournis dans les 30 jours qui suivront la date à laquelle le CEPMB lui enverra une demande suite à la réception d’une plainte puis, pour les deux prochaines années, dans les 30 jours suivant la fin du semestre de rapport. Il devra également tenir à jour les renseignements exigés sur le formulaire 2 depuis la date de la première vente de son médicament au Canada au cas où le CEPMB recevrait une plainte et lui demanderait les mêmes renseignements pour ces périodes.
- Le breveté ou l’ancien breveté qui choisit de ne pas soumettre au CEPMB les renseignements exigés sur le formulaire 2 pour un médicament qu’il vend entre la date à laquelle la demande de brevet est devenue accessible au public et l’octroi du brevet, doit garder ses données à jour en prévision du jour où il devra soumettre son rapport, soit suivant l’octroi du brevet.
7.3 Tous les renseignements énumérés dans le paragraphe 6.2 doivent être soumis au CEPMB sur les formulaires électroniques pouvant être téléchargés depuis le site Web du CEPMB. Vous trouverez ces formulaires sous la rubrique « Loi, Règlement et Lignes directrices, puis sous « Guide du breveté ». Le rapport rempli doit être transmis par courriel au CEPMB à l’adresse spécifiée dans son site Web.
7.4 Les rapports électroniques soumis au CEPMB doivent porter la signature numérique de la personne dûment autorisée à certifier l’exactitude des renseignements fournis.
8. Sanctions pour défaut de présenter les rapports exigés aux finsde la réglementation des prix
8.1 Tout élément de preuve établissant que le breveté n’a pas signifié son « Avis d’intention de vendre un médicament breveté sur le marché canadien » en application du paragraphe 82(1) de la Loi sera porté à l’attention du président du Conseil qui décidera s’il y a lieu d’émettre une ordonnance à l’encontre du breveté pour l’obliger à soumettre au Conseil son « Avis d’intention ».
8.2 À défaut du breveté ou de l’ancien breveté de soumettre dans les délais impartis un rapport ou tous ses rapports sur le formulaire 1 ou sur le formulaire 2 pour une ou pour plusieurs périodes de rapport, le personnel du Conseil lui fera savoir par écrit qu’il est en défaut de soumettre son ou ses rapports et lui accordera un délai de grâce de sept jours commençant à la date d’envoi de la lettre du personnel du Conseil. Si le breveté ou l’ancien breveté néglige de fournir les renseignements demandés dans les délais impartis, le personnel du Conseil recommandera au président du Conseil sans autre préavis de rendre une ordonnance en vertu de l’article 81 de la Loi afin d’obliger le breveté ou l’ancien breveté de lui fournir les renseignements demandés dans le délai dicté dans l’ordonnance.
8.3 Si le président du Conseil ou si le Conseil arrive à la conclusion que le breveté a sciemment omis de présenter les rapports exigés en vertu des articles 80, 81 ou 82 de la Loi, ou suivant l’émission d’une ordonnance du Conseil, le président ou le Conseil saisira le procureur général du Canada de la situation afin qu’il engage s’il y a lieu des poursuites judiciaires en vertu de l’alinéa 76.1(1) de la Loi.
8.4 En application de l’article 99 de la Loi, les ordonnances rendues par le Conseil peuvent être assimilées à des ordonnances de la Cour fédérale ou d’une cour supérieure et, le cas échéant, leur exécution sera effectuée suivant les mêmes modalités.
9. Protection du caractère confidentiel des renseignements fournis dans le cadredu processus de réglementation du prix
9.1 En vertu du paragraphe 87(1) de la Loi, sauf les exceptions mentionnées ci-après, tous les renseignements ou documents fournis au Conseil en application des articles 80, 81 ou 82 de la Loi ou dans le cadre d’une audience tenue en vertu de l’article 83 sont protégés et ne peuvent être communiqués sans l’autorisation de la personne qui les a fournis, sauf s’ils ont été divulgués dans le cadre d’une audience publique tenue en vertu de l’article 83.
9.2 Tout renseignement appartenant au domaine public qu’un breveté ou un ancien breveté fournit au CEPMB ne sera pas considéré protégé en vertu du paragraphe 87(1) de la Loi. Au nombre de ces renseignements citons les prix départ-usine au Canada et dans les pays de comparaison d’un médicament breveté qui sont disponibles au grand public (Formulaire 2, section 5), les monographies du produit disponible au grand public, les résultats des essais cliniques et les lignes directrices sur la pratique.
9.3 La protection accordée aux renseignements en vertu du paragraphe 87(1) ne s’applique pas aux renseignements et aux documents colligés par le CEPMB, incluant toute analyse de ces renseignements effectuée par le personnel du Conseil. 9.4 Le paragraphe 86(1) de la Loi donne au Conseil le pouvoir discrétionnaire de tenir à huis clos en tout ou en partie une audience si le Conseil est convaincu, à la suite d’observations faites par l’intéressé que la divulgation des renseignements ou documents en cause dans le cadre d’une audience publique causerait directement au breveté un préjudice réel et sérieux .
9.5 Les renseignements concernant le statut de l’examen du prix effectué par le personnel du Conseil, y compris du statut de conformité des brevetés, ne sont pas protégés en vertu du paragraphe 87(1) et en conséquence peuvent être rendus publics. En conséquence, le CEPMB publiera des rapports sommaires des résultats de l’examen du prix de toutes les nouvelles substances actives. Le CEPMB est également en droit de publier les résultats d’autres examens.
9.6 En vertu du paragraphe 87(2) de la Loi, le CEPMB peut communiquer tout renseignement ou document fourni au CEPMB en application des articles 80, 81 ou 82 de la Loi ou dans le cadre d’une audience tenue en vertu de l’article 83 à quiconque est chargé, sous sa responsabilité, de l’application de la Loi ainsi qu’au ministre de l’Industrie ou tout autre ministre désignée par règlement, ou à un ministre provincial responsable de la santé ou à tel de leurs fonctionnaires à seule fin de leur permettre de présenter leurs observations au Conseil concernant l’audience tenue en vertu de l’article 83.
Partie II – Les politiques
Introduction
Le Conseil adopte de temps à autre des politiques afin que ses intervenants connaissent les principes qu’il applique dans l’exercice de son mandat. Nous vous présentons ci-après les principales politiques dont s’est doté le Conseil au fil des années. Même si le Conseil n’est pas lié par ses politiques, elles lui permettent de faire preuve de transparence et de cohésion auprès de ses intervenants.
1. Politique à l’égard du brevet en instance
1.1 Lorsqu’un médicament en instance d’être breveté est vendu sur un marché canadien, le CEPMB fera l’examen du prix au moment de l’octroi du brevet. Le prix faisant alors l’objet de l’examen est le prix à la date de la première vente du médicament ou la date à laquelle la demande de brevet est devenue accessible au public, soit la dernière de ces deux éventualités. Lorsque le brevet a été octroyé, la compétence du Conseil est rétroactive au prix auquel le médicament a été vendu alors que la demande de brevet était devenu accessible au public du fait que la partie qui vend le médicament a tiré un avantage du brevet au cours de cette période au même titre que le ferait un « breveté » en vertu du paragraphe 79(1) de la Loi.
2. Politique à l’égard du brevet cédé au domaine public
2.1 Le CEPMB continue d’exercer sa compétence sur le prix de vente d’un médicament breveté sur un marché canadien dont le brevet a été cédé au domaine public et ce, tant que le brevet n’a pas été annulé ou cédé de la façon dont le prévoit la Loi sinon tant que le brevet ne sera pas arrivé à échéance. La Loi, qui constitue le mécanisme en vertu duquel l’État attribue des brevets et confère aux brevetés des droits et des avantages pendant la durée du brevet, ne reconnaît pas expressément la cession des brevets au domaine public comme mécanisme pour mettre fin aux droits que confère un brevet avant sa date d’échéance.
3. Politique concernant le sens donné au mot « médicament »
3.1 Un médicament s´entend de toute substance ou mélange de substances fabriqué par quelque moyen biologique, chimique ou autre et qui est appliqué ou administré in vivo à des humains ou à des animaux pour faciliter le diagnostic, le traitement, l´atténuation ou la prévention d´une maladie, de symptômes, de troubles ou d´états physiques anormaux ou, encore, qui modifie des fonctions organiques chez les humains ou chez les animaux, sans égard à son mode d´administration.
3.2 Il est entendu que la définition du mot « médicament » vise également les vaccins, les préparations topiques, les anesthésiques et les produits de diagnostic utilisés in vitro, sans égard au mode d´administration (par ex. administration transdermique, gélule, solution injectable, inhalation, etc.). Cette définition exclut les appareils médicaux, les produits diagnostiques et les désinfectants non utilisés in vivo.
4. Politique à l’égard de l’unité d’examen du prix
4.1 Le CEPMB fait l’examen du prix moyen de chaque concentration de chaque forme posologique finale de chaque produit médicamenteux breveté vendu au Canada, nommément :
- des médicaments auxquels Santé Canada a attribué un numéro d’identification de drogue (DIN)
- des médicaments vendus au titre du Programme d’accès spécial
- des médicaments distribués en vertu d’une demande de participation à des essais cliniques
- des drogues nouvelles de recherche
4.2 Dans le présent document, l’expression « produit médicamenteux » désigne chaque concentration de chaque forme posologique finale de chaque médicament breveté.
4.3 Le prix moyen d’un produit médicamenteux correspond généralement au prix de l’unité dans laquelle le médicament est vendu (par ex. comprimé, millilitre, inhalateur, etc.) arrondi à la quatrième décimale.
5. Politique appliquée lorsque le prix semble excessif
5.1 Le prix d’un produit médicamenteux semble excessif dans les situations suivantes :
- lorsque, pour la période de lancement, le prix de transaction moyen national du produit est plus élevé que son prix moyen maximal potentiel ou, pour les périodes de rapport subséquentes, que le prix moyen non excessif national
- lorsque, pour la période de lancement, le prix de transaction moyen du marché est plus élevé que le prix moyen maximal potentiel ou, pour les périodes de rapport subséquentes, que le prix moyen non excessif du marché respectif.
5.2 Lorsque le prix de transaction moyen national est plus élevé que le prix moyen maximal potentiel ou que le prix moyen non excessif national, mais dans une mesure trop minime pour justifier une enquête (appendice 11), le personnel du Conseil informera le breveté de la situation et mention sera faite dans le site Web du CEPMB que le prix du produit médicamenteux brevet « semble excessif ». Le breveté devra alors réduire le prix de son produit et rembourser les recettes excessives qu’il a tirées de la vente de son produit à un prix excessif (voir dans la section 7 la politique du CEPMB concernant le remboursement des recettes excessives).
5.3 Lorsque le prix de transaction moyen national dépasse le prix moyen maximal potentiel ou le prix moyen non excessif national dans une mesure qui justifie la tenue d’une enquête, le personnel du Conseil informera le breveté qu’il a engagé une enquête sur le prix de son produit et le produit sera classé « Sous enquête » (chapitre 3 « Enquêtes »).
6. Politique sur l’utilisation des produits médicamenteux brevetés et non brevetésdans les tests appliqués aux prix
6.1 Le personnel du Conseil peut exclure des comparaisons tout produit médicamenteux breveté ou non breveté retenu pour la comparaison s’il a des raisons de croire que le produit est vendu au Canada à un prix excessif.
6.2 Les produits clés utilisés pour les comparaisons seront généralement soumis aux tests appliqués au prix décrit dans les Lignes directrices.
7. Politique à l’égard du remboursement des recettes excessives
7.1 Tel que mentionné dans la section 6 du cadre législatif, le Conseil peut permettre au breveté de rembourser le montant estimé des recettes excessives qu’il a tirées de la vente de son médicament à un prix excessif et ce, de l’une ou l’autre des façons suivantes : (1) en réduisant le prix de son médicament ou, encore, le prix d’un autre médicament breveté qu’il vend au Canada, ou (2) en remettant à Sa Majesté du chef du Canada le montant estimé des recettes excessives qu’il a encaissées.
7.2 Lorsque les recettes excessives sont remboursées au moyen d’une réduction de prix, le prix moyen du produit médicamenteux breveté faisant l’objet du remboursement ne sera considéré avoir été réduit que s’il est moins élevé que le prix moyen non excessif de l’année précédente. Le renoncement à l’augmentation autorisée du prix du produit ne sera pas une mesure de remboursement des recettes excessives acceptable.
7.3 Le remboursement en trop des recettes excessives ne peut créer un compte déficitaire.
Partie III – Lignes directrices et procédures
Préface
Les Lignes directrices et les procédures sont les directives que le Conseil donne aux brevetés et au personnel du Conseil afin que les prix des produits médicamenteux brevetés soient conformes à la Loi sur les brevets et au Règlement sur les médicaments brevetés. Il importe de noter que les brevetés et le Conseil ne sont pas liés par les Lignes directrices dans le cadre d’une audience.
Les Lignes directrices sont présentées de la façon suivante :
Chapitre 1 – Processus d’examen scientifique : Processus fondé sur les données cliniques qui évalue la mesure dans laquelle un nouveau produit médicamenteux breveté offre des bienfaits thérapeutiques par rapport aux produits médicamenteux existants. À la lumière des résultats obtenus, une recommandation est formulée concernant les produits médicamenteux qui se prêtent à une comparaison ainsi que leurs régimes posologiques comparables.
Chapitre 2 – Processus d’examen du prix : C’est à la lumière du niveau d’amélioration thérapeutique du produit médicamenteux breveté qu’est déterminé le prix moyen maximal potentiel pour la période de lancement. Pour les périodes de rapport subséquentes, le prix d’un produit médicamenteux breveté existant est soumis aux tests de prix pertinents aux fins de déterminer son prix moyen non excessif national et ses prix moyens non excessifs du marché.
Chapitre 3 – Enquêtes : L’approche utilisée et les procédures suivies lorsque le prix d’un produit médicamenteux semble être excessif dans une mesure qui justifie la tenue d’une enquête (appendice 11).
Appendices : Les appendices constituent une partie intégrante des Lignes directrices.
Chapitre 1 – Le processus d’examen scientifique
1. Introduction
1.1 Le processus d’examen scientifique du CEPMB se fonde sur la preuve clinique. C’est en fonction des résultats de l’examen scientifique qu’est recommandé le niveau d’amélioration thérapeutique du produit médicamenteux breveté.
1.2 Tous les nouveaux produits médicamenteux brevetés (incluant les produits ayant obtenu leur Avis de conformité, les produits distribués par le truchement du Programme spécial d’accès, les produits faisant l’objet d’une demande d’essai clinique et les nouvelles drogues de recherche) sont soumis à l’examen scientifique conformément aux Lignes directrices et aux procédures présentées dans le présent chapitre.
2. Sources des données scientifiques
2.1 Pour l’évaluation scientifique d’un nouveau produit médicamenteux breveté, le personnel du Conseil utilise les renseignements provenant des sources suivantes :
- Présentation du breveté – Le breveté est invité à soumettre au personnel du Conseil une brève présentation (voir l’appendice 1) dans laquelle il motive sa proposition quant au niveau d’amélioration de bienfaits thérapeutiques de son produit médicamenteux, la sélection des produits médicamenteux qui se prêtent à la comparaison avec son produit et des régimes posologiques comparables.
- Rapports de recherches effectuées par un Centre d’information sur le médicament (CIM) – Le personnel du Conseil obtient des données scientifiques auprès de CIM, entre autres des rapports sur les essais cliniques, les lignes directrices pour la pratique clinique, etc. La base de l’examen qu’effectue le CIM est la monographie du produit ou, si l’Avis de conformité n’a pas encore été attribué au produit, les renseignements que contient généralement une monographie de produit.
- Recherche effectuée par le personnel du Conseil – Le personnel du Conseil peut également mettre à jour ses recherches et consulter d’autres sources pour compléter ses données ainsi que les éléments de preuve fournis par le breveté ou par le CIM.
- Résultats des recherches des membres du GCMUH – Les membres du Groupe consultatif sur les médicaments pour usage humain peuvent également effectuer leur propre recherche et ainsi compléter les éléments de preuve fournis par les brevetés, par le CIM et par les membres du personnel.
3. Groupe consultatif sur les médicaments pour usage humain
3.1 Le Groupe consultatif sur les médicaments pour usage humain (GCMUH) met son expertise au service du personnel du Conseil en effectuant des examens scientifiques. Le GCMUH est chargé des fonctions suivantes :
- il évalue les renseignements scientifiques obtenus auprès des sources mentionnées dans le paragraphe 2
- lorsque nécessaire, il sollicite l’opinion d’autres experts
- il recommande le niveau d’amélioration thérapeutiques et identifie les produits médicamenteux qui se prêtent aux comparaisons ainsi que, lorsque possible, les régimes posologiques
- dans les éléments de preuve, il relève les incertitudes qui sont susceptibles d’avoir une incidence sur l’analyse et, par ricochet, sur les recommandations qui en découleront.
3.2 En règle générale, les nouveaux produits médicamenteux brevetés sont soumis à l’examen du GCMUH. Toutefois, les nouveaux produits médicamenteux brevetés nommés ci-après ne sont pas soumis à l’examen du GCMUH à moins que le breveté n’allègue dans sa présentation que son produit offre de plus grands bienfaits thérapeutiques que les médicaments existants :
- le nouveau produit médicamenteux breveté représente un nouveau DIN d’une forme posologique d’un produit médicamenteux existant ou, encore, un nouveau DIN d’une autre forme posologique du produit médicamenteux comparable à la forme posologique existante (selon la description donnée à l’appendice 2) qui a la même indication ou la même utilisation que le DIN existant, ou
- le nouveau produit médicamenteux breveté est une combinaison de produits médicamenteux dont les différentes composantes sont vendues au Canada pour la même indication ou pour la même utilisation, ou
- Santé Canada considère le nouveau médicament générique breveté bioéquivalent au produit médicamenteux de marque de référence vendu au Canada, ou
- Le nouveau produit médicamenteux générique breveté est une version d’un médicament de marque existant vendu au Canada produite sous licence.
Procédures :
3.3 Les membres du GCMUH ont une expertise reconnue en pharmacothérapie ainsi qu’une solide expérience en méthodologie de recherche scientifique, en analyse statistique et en évaluation de nouveaux produits médicamenteux.
3.4 Le GCMUH et ses membres ne rencontrent pas les brevetés.
3.5 L’identité des membres du GCMUH est divulguée dans le site Web du CEPMB.
3.6 Les dates des réunions du GCMUH sont annoncées dans le site Web du CEPMB.
3.7 À la demande d’un breveté, un nouveau produit médicamenteux breveté est soumis à la consultation du GCMUH aux fins d’obtenir ses conseils avant que le produit ne soit offert en vente au Canada et (ou) avant l’octroi d’un brevet.
3.8 Pour que le nouveau produit médicamenteux breveté puisse être soumis à l’examen du GCMUH, la présentation du breveté doit contenir les différents éléments mentionnés dans l’appendice 1 et être soumise au GCMUH au moins deux mois avant la réunion où le Groupe doit faire l’évaluation du produit.
3.9 Si le GCMUH est appelé à se pencher sur un nombre important de soumissions au cours d’une de ses réunions, les présentations seront évalués suivant l’ordre de priorité suivant :
- produits médicamenteux brevetés et vendus au Canada
- produits médicamenteux brevetés et devant être vendus au Canada à brève échéance
- produits médicamenteux brevetés, mais pas encore vendus sur le marché canadien
- produits médicamenteux qui ne sont pas brevetés, mais qui sont vendus au Canada
- produits médicamenteux qui ne sont pas brevetés et qui ne sont pas non plus vendus au Canada.
3.10 Le breveté sera informé de la date de la réunion du GCMUH au cours de laquelle sa soumission sera évaluée.
3.11 Dans son rapport, le GCMUH formulera ses recommandations quant à la mesure dans laquelle le produit médicamenteux offre de plus grands bienfaits thérapeutiques par rapport aux produits médicamenteux existants et proposera sa sélection de produits médicamenteux et des régimes posologiques qui se prêtent à la comparaison. Le GCMUH décrira également dans son rapport la façon dont il a appliqué les facteurs primaires et secondaires (voir ci-après la section 6) ainsi que les éléments de preuve (voir ci-après la section 7) sur lesquels il fonde sa recommandation.
3.12 Le breveté recevra un exemplaire du rapport du GCMUH.
4. Détermination de l’indication/utilisation principale du nouveau produit médicamenteux breveté
4.1 L’indication principale approuvée (ou l’indication proposée lorsque le produit n’a pas encore obtenu son Avis de conformité) doit être déterminée pour que puisse être évalué le niveau d’amélioration des bienfaits thérapeutiques d’un nouveau produit médicamenteux breveté utilisé pour de multiples indications approuvées/utilisations.
Procédures :
4.2 Le niveau d’amélioration des bienfaits thérapeutiques d’un nouveau produit médicamenteux breveté utilisé pour différentes indications approuvées est déterminé à l’aide de l’indication ou de l’utilisation approuvée, soit l’indication ou l’utilisation qui offre les plus grands bienfaits thérapeutiques par rapport aux autres thérapies administrées pour la même indication/utilisation à un segment assez important de patients. Ainsi, les conditions médicales rares ou les maladies peu courantes (qui ont donc une faible incidence ou une faible prévalence au Canada) ne sont pas prises en compte.
4.3 L’indication approuvée ou l’utilisation sera considérée « l’indication principale » aux fins de la sélection des produits médicamenteux pour les différentes comparaisons de prix.
4.4 S’il n’existe aucune indication approuvée ou utilisation pour laquelle le nouveau produit médicamenteux breveté offre une amélioration marquée des bienfaits thérapeutiques par rapport aux produits existants, c’est l’indication approuvée ou l’utilisation présentant le plus grand potentiel de ventes qui déterminera le niveau d’amélioration des bienfaits thérapeutiques par rapport aux produits existants ainsi que la sélection des produits qui seront utilisés pour les différentes comparaisons de prix.
4.5 Le potentiel des ventes peut être évalué à l’aide de différents facteurs, dont les habitudes d’ordonnance des médecins (lorsque celles-ci sont connues), les données épidémiologiques (incidence et prévalence de la condition au Canada) et les habitudes d’ordonnance dans les autres pays.
5. Niveau d’amélioration des bienfaits thérapeutiques
5.1 Dans ses recommandations concernant le niveau d’amélioration des bienfaits thérapeutiques, le GCMUH utilise les définitions suivantes :
Médicaments constituant une découverte : Premier médicament de sa catégorie disponible sur le marché canadien qui traite avec efficacité une maladie ou une condition particulière.
Médicaments constituant une amélioration importante : Produit médicamenteux qui offre des bienfaits thérapeutiques largement plus importants à ceux des produits existants offerts sur le marché canadien.
Médicaments constituant une amélioration modeste : Produit médicamenteux qui par rapport aux produits existants vendus sur le marché canadien offre des bienfaits thérapeutiques modestement meilleurs.
Médicaments constituant une amélioration minime ou nulle : Produit médicamenteux qui par rapport aux produits existants vendus sur le marché canadien offre peu ou aucun bienfait thérapeutique supplémentaire.
6. Facteurs à l’appui de la recommandation du niveau d’amélioration thérapeutique
6.1.1 Les facteurs sousmentionnés justifient la recommandation du niveau d’amélioration des bienfaits thérapeutiques d’un produit médicamenteux :
Facteurs principaux
- plus grande efficacité
- moins grande incidence ou intensité des effets indésirables
Facteurs secondaires
- voie d’administration
- facilité d’utilisation pour le patient
- plus grande conformité donnant lieu à une plus grande efficacité thérapeutique
- facilité au niveau de la prestation des soins
- temps nécessaire pour obtenir l’effet thérapeutique optimal
- durée du traitement habituel
- taux de succès
- pourcentage d’efficacité sur la population visée
- prévention d’une incapacité/économies
6.2 Les facteurs principaux ont une plus grande incidence sur la recommandation après quoi sont prises en compte les améliorations attribuables aux facteurs secondaires.
6.3 La recommandation concernant le niveau d’amélioration des bienfaits thérapeutiques d’un nouveau produit médicamenteux breveté ne tiendra pas compte des facteurs sousmentionnés à moins qu’ils ne favorisent une plus grande efficacité et/ou une réduction de l’incidence ou de l’intensité des effets indésirables :
- mécanisme d’action du produit médicamenteux
- nouvelle entité chimique
- autre profil pharmacocinétique
Procédures :
6.4 Les facteurs principaux seront pris en compte pour déterminer si le nouveau produit médicamenteux breveté constitue une découverte ou s’il constitue une amélioration importante, modeste ou minime/nulle par rapport aux autres produits médicamenteux vendus sur le marché canadien.
6.5 Les facteurs secondaires seront considérés en deuxième lieu. Le GCMUH pondérera ces facteurs selon les éléments de preuve qui lui sont soumis et selon également un jugement clinique raisonnable. Ces facteurs peuvent mener au classement du produit dans une catégorie constituant tout au plus une amélioration modeste.
7. Méthodologie d’évaluation du niveau d’amélioration des bienfaits thérapeutiques
7.1 Une approche qui se fonde sur la preuve clinique sera utilisée pour évaluer le nouveau produit médicamenteux breveté sous examen selon la hiérarchie de la preuve établie par le Oxford Centre for Evidence-Based Medicine (appendice 1).
Procédures :
7.2 Le GCMUH évalue d’une façon critique les éléments de preuve sous les angles de leur validité, de leur incidence et de leur facilité d’application. Les éléments de preuve du niveau 1 auront préséance sur ceux des autres niveaux dans la recommandation du niveau d’amélioration des bienfaits thérapeutique et des médicaments qui se prêtent aux comparaisons.
7.3 L’efficacité relative d’un nouveau produit médicamenteux breveté étant souvent incertaine, le GCMUH utilise autant que possible les éléments de preuve du premier niveau lorsqu’il évalue de nouveaux produits médicamenteux brevetés constituant une découverte ou une amélioration importante par rapport aux autres produits médicamenteux vendus au Canada.
7.4 Selon le cas, le GCMUH peut considérer des éléments de preuve d’autres niveaux lorsqu’il évalue les facteurs secondaires qui sont susceptibles d’avoir une incidence sur le classement selon le niveau d’amélioration des bienfaits thérapeutiques.
8. Sélection des produits médicamenteux et des régimes posologiques aux fins de comparaisons
Produits médicamenteux aux fins de comparaisons du prix
8.1 Pour la sélection des produits médicamenteux aux fins de comparaisons, le GCMUH utilise le système de classification Anatomique Thérapeutique Chimique (ATC) du Centre collaborateur de l’Organisation mondiale de la santé (OMS) pour la méthodologie sur l’établissement des statistiques concernant les produits médicamenteux.
8.2 Pour la comparaison, les substances chimiques dans le sous-groupe de la classification ATC qui précède la substance active simple, soit généralement le 4e sous-groupe. Le GCMUH peut également sélectionner les substances dans le sous-groupe qui précède immédiatement le 4e sous-groupe ou dans un autre sous-groupe. Dans certains cas, il pourra sélectionner la substance chimique dans le sous-groupe qui correspond à la substance chimique simple, soit le 5e sous-groupe.
8.3 Le GCMUH peut exclure de sa comparaison une substance chimique du même groupe ATC que le nouveau produit médicamenteux breveté sous examen s’il estime que ce produit ne se prête pas à la comparaison. Par exemple, les produits médicamenteux dont l’indication principale/ l’utilisation n’est pas la même que celle de l’indication principale/l’utilisation du nouveau produit médicamenteux sous examen peuvent être exclus de la comparaison.
Procédures :
8.4 Le GCMUH fait la sélection des différents produits médicamenteux qui seront utilisés pour la comparaison parmi les produits qui ont la même indication approuvée/utilisation que le nouveau produit médicamenteux breveté sous examen.
Découverte
8.5 Lorsque le nouveau produit médicamenteux breveté constitue une découverte, le GCMUH ne recommande aucun produit pour la comparaison puisqu’il n’existe en toute logique aucun produit semblable sur le marché. En effet, le nouveau produit est par définition le premier produit de sa catégorie qui réussit à traiter avec efficacité une maladie ou une condition donnée.
Amélioration importante
8.6 Lorsque le nouveau produit médicamenteux breveté constitue une amélioration importante des bienfaits thérapeutiques par rapport aux produits existants, le GCMUH identifiera les produits médicamenteux ayant la même indication approuvée ou la même utilisation que le produit par rapport auquel le produit sous examen constitue une amélioration importante.
Amélioration modeste
8.7 Lorsque le nouveau produit médicamenteux breveté constitue une amélioration modeste des bienfaits thérapeutiques par rapport aux produits existants, le GCMUH identifiera les produits ayant la même indication approuvée ou la même utilisation que le produit sous examen et par rapport auxquels le nouveau produit médicamenteux breveté constitue une amélioration modeste.
Amélioration minime ou nulle
8.8 Tout produit médicamenteux qui n’est pas considéré comme une découverte, une amélioration importante ou une amélioration minime sera automatiquement classé dans la catégorie des produits médicamenteux qui constituent une amélioration minime ou nulle par rapport aux produits existants.
8.9 Pour qu’un nouveau produit médicamenteux breveté soit classé dans la catégorie des produits médicamenteux constituant une amélioration minime ou nulle, le GCMUH tentera en premier lieu d’identifier les médicaments qui se prêtent à la comparaison à l’aide des facteurs primaires et secondaires présentés dans le paragraphe 6.1 du présent chapitre. Ces médicaments devront avoir la même indication approuvée/utilisation que le nouveau produit médicamenteux sous examen.
8.10 Si le GCMUH n’arrive pas à identifier de produits qui se prêtent à une comparaison avec le produit médicamenteux sous examen, il identifiera les produits qui sont réputés le mieux traiter l’indication approuvée ou l’utilisation en référence aux facteurs primaires et secondaires présentés dans le paragraphe 6.1 du présent document.
8.11 La sélection des produits pour fins de comparaisons avec celui d’un nouveau produit médicamenteux breveté qui est en fait une nouvelle présentation de la même entité chimique qu’un autre produit médicamenteux ayant la même forme posologique ou une forme posologique comparable (appendice 2), la même indication ou la même utilisation ainsi que le même régime posologique sera limitée à l’entité chimique du nouveau produit médicamenteux breveté sous examen ainsi qu’à la même forme posologique ou à des formes posologiques comparables (appendice 2) à moins que le breveté n’ait soumis une présentation dans laquelle il allègue une amélioration des bienfaits thérapeutiques et que cette allégation soit confirmée par le GCMUH.
8.12 Autrement dit, la sélection des produits médicamenteux de comparaison pour un nouveau produit médicamenteux breveté qui est une nouvelle présentation de la même entité chimique qu’un autre produit médicamenteux ayant la même forme posologique ou une forme posologique comparable (appendice 2) ainsi que la même indication ou utilisation, mais un régime posologique fort différent, sera limitée à la même entité chimique présentée dans la même forme posologique ou dans une forme posologique comparable (appendice 2) à moins que le breveté n’allègue dans une présentation que son produit offre de plus grands bienfaits thérapeutiques que les produits existants et que le GCMUH confirme que le nouveau produit médicamenteux breveté offre effectivement de plus grands bienfaits thérapeutiques.
8.13 La sélection des produits médicamenteux qui se prêtent à une comparaison avec un nouveau produit médicamenteux combiné où chaque élément du produit est vendu au Canada pour la même indication ou pour la même utilisation sera limitée aux différents éléments constituant le produit médicamenteux à moins que le breveté ne soumette une présentation selon laquelle son nouveau produit offre de plus grands bienfaits thérapeutiques que les médicaments existants et que le GCMUH confirme que le produit médicamenteux breveté sous examen offre une telle amélioration.
8.14 La sélection des produits médicamenteux qui se prêtent à une comparaison avec un nouveau produit médicamenteux breveté générique bioéquivalent au produit de marque vendu au Canada ou fabriqué sous licence sera limitée au produit médicamenteux de marque. Régimes posologiques comparables
8.15 Le régime posologique recommandé pour la comparaison ne doit pas être plus généreux que le régime posologique le plus généreux recommandé dans la monographie du produit (ou renseignements de semblable nature lorsque l’Avis de conformité n’a pas été attribué au produit médicamenteux) et tiendra compte des variables cliniques pertinentes. La concentration la plus appropriée du produit médicamenteux sera également choisie pour le régime posologique recommandé.
8.16 En règle générale, un régime posologique pour un traitement sera recommandé lorsque l’indication vise une maladie ou une condition grave alors que pour des conditions chroniques, c’est un régime posologique quotidien (dose de maintien) qui sera recommandé.
9. Dispositions visant les produits médicamenteux en vente libre et les produits médicamenteux pour usage vétérinaire
9.1 Conformément au Règlement et aux dispositions concernant la présentation de rapport présentées dans la Partie I – Le cadre juridique, le CEPMB n’effectuera un examen scientifique des produits médicamenteux en vente libre et des produits médicamenteux pour usage vétérinaire que suite à la réception d’une plainte concernant le prix du produit médicamenteux breveté.
Procédures :
9.2 Sur réception d’une plainte, le CEPMB effectuera l’examen scientifique du produit médicamenteux breveté en vente libre ou du produit médicamenteux breveté pour usage vétérinaire de la même façon qu’il effectue l’examen scientifique de tous les autres produits médicamenteux brevetés, tel que décrit dans le présent chapitre.
9.3 Sur réception d’une plainte concernant le prix d’un produit médicamenteux breveté en vente libre, les données scientifiques pertinentes seront examinées par le GCMUH afin qu’il recommande le classement du produit médicamenteux selon la mesure dans laquelle il offre de plus grands bienfaits thérapeutiques, les produits médicamenteux et les régimes posologiques comparables.
9.4 S’il reçoit une plainte concernant le prix d’un produit médicamenteux breveté pour usage vétérinaire, le CEPMB constituera un Groupe consultatif sur les médicaments pour usage vétérinaire aux fins qu’il formule des recommandations concernant le classement du produit selon la mesure dans laquelle il offre de plus grands bienfaits thérapeutiques, les produits médicamenteux et les régimes posologiques qui se prêtent à une comparaison.
Chapitre 2 – Le processus d’examen du prix
1. Introduction
1.1 Les objectifs de l’examen du prix sont les suivants :
- Déterminer le prix moyen maximal potentiel auquel le nouveau produit médicamenteux breveté peut être vendu au cours de sa période de lancement; et
- Déterminer si le prix d’un produit médicamenteux breveté existant est ou non excessif.
2. Examen des prix auxquels les nouveaux produits médicamenteux brevetés sont vendus durant la période de lancement
Introduction
2.1 Le choix du test qui sera appliqué au prix de lancement d’un nouveau produit médicamenteux breveté varie selon le classement du produit selon le niveau d’amélioration thérapeutique recommandé à la lumière des résultats de l’examen scientifique (chapitre 1). Vous trouverez dans l’appendice 8 une description détaillée de la façon dont les tests seront appliqués aux prix selon le niveau d’amélioration thérapeutique.
2.2 Le CEPMB se réserve de droit de faire l’examen du prix de tout produit médicamenteux breveté vendu sur un marché au Canada (y compris pour une catégorie de clients et d’une province ou d’un territoire).
Test du prix international le plus élevé
2.3 Par dérogation aux paragraphes 2.4 à 2.8, le prix moyen maximal potentiel auquel un nouveau produit médicamenteux breveté peut être vendu au cours de sa période de lancement ne doit pas être plus élevé que le prix obtenu avec le test du prix international le plus élevé. (appendice 6).
Produits médicamenteux constituant une découverte
2.4 Sous réserve du paragraphe 2.3, le prix de lancement d’un nouveau produit médicamenteux constituant une découverte sera jugé excessif si son prix de transaction moyen national ou son prix de transaction moyen du marché est plus élevé que le prix moyen maximal potentiel pour la période de lancement établi au moyen de la Comparaison avec le prix international médian (appendice 5).
Produits médicamenteux constituant une amélioration importante
2.5 Sous réserve du paragraphe 2.3, le prix de lancement d’un nouveau produit médicamenteux breveté qui constitue une amélioration importante sera considéré excessif si son prix de transaction moyen national ou son prix de transaction moyen du marché est plus élevé que le prix moyen maximal potentiel pour la période de lancement, lequel correspondra au plus élevé des prix calculés comme suit :
- (a) le prix non excessif le plus élevé calculé de la façon expliquée dans le paragraphe 8.6 du chapitre 1 soit avec la comparaison du prix selon la catégorie thérapeutique (appendice 3), et
- (b) la médiane des prix auquel le produit est vendu dans les différents pays de comparaison (appendice 5).
Produits médicamenteux constituant une amélioration modeste
2.6 Le prix de lancement d’un nouveau produit médicamenteux qui constitue une amélioration modeste sera considéré excessif si son prix de transaction moyen national ou son prix de transaction moyen du marché est plus élevé que son prix moyen maximal potentiel pour la période de lancement. Ce dernier prix correspondra au plus élevé des prix obtenus avec les tests suivants :
- (a) Le point médian entre le prix obtenu dans l’alinéa (b) et le prix médian international obtenu selon le test de la comparaison du prix international médian (appendice 5); et
- (b) Le prix non excessif le plus élevé de tous les produits médicamenteux identifiés calculé de la façon expliquée dans le paragraphe 8.7 du chapitre 1, soit la comparaison du prix selon la catégorie thérapeutique (appendice 3).
2.7 Lorsqu’il n’est pas possible d’effectuer une comparaison selon la catégorie thérapeutique, le prix de lancement d’un nouveau produit médicamenteux constituant une amélioration modeste sera jugé excessif si le prix de transaction moyen national ou le prix de transaction moyen du marché est plus élevé que le prix médian international obtenu avec la comparaison du prix international médian (appendice 5). Telle situation peut se produire lorsque le GCMUH n’arrive pas à trouver des régimes posologiques comparables pour tous les produits identifiés conformément au paragraphe 8.7 du chapitre 1 ou lorsque les prix de ces produits médicamenteux semblent excessifs.
Produit médicamenteux constituant une amélioration minime ou nulle
2.8 Le prix de lancement d’un nouveau produit médicamenteux breveté qui constitue une amélioration minime ou nulle par rapport aux produits médicamenteux existants sera considéré excessif si son prix de transaction moyen national ou son prix de transaction moyen du marché est plus élevé que son prix moyen maximal potentiel. Ce dernier prix correspond au prix non excessif le plus élevé de tous les produits médicamenteux retenus pour la comparaison du prix selon la catégorie thérapeutique (appendice 3) et identifiés de la façon décrite dans le paragraphe 8.9 du chapitre 1.
2.9 Le GCMUH peut arriver à la conclusion que le nouveau produit médicamenteux qui constitue une amélioration minime ou nulle ne compte aucun produit médicamenteux qui se prête à une comparaison. Dans un tel cas aussi exceptionnel, le prix de lancement du nouveau produit médicamenteux breveté sera considéré excessif si son prix de transaction moyen national ou son prix de transaction moyen du marché est plus élevé que le moindre des prix suivants :
- (a) le prix non excessif le plus bas des produits médicamenteux offrant de plus grands bienfaits thérapeutiques identifiés au moyen d’une comparaison du prix selon la catégorie thérapeutique de la façon décrite dans le paragraphe 8.10 du chapitre 1.
- (b) Le prix médian international calculé à l’aide de la comparaison avec le prix international médian (appendice 5).
2.10 S’il n’est pas possible d’effectuer une comparaison selon la catégorie thérapeutique, le prix de lancement du nouveau produit médicamenteux qui constitue une amélioration minime ou nulle par rapport aux produits existants sera considéré excessif si son prix de transaction moyen national ou un de ses prix de transaction moyens du marché est plus élevé que le prix international médian calculé à l’aide de la comparaison du prix avec le prix international médian (appendice 5). Telle situation peut se produire lorsque le GCMUH n’arrive pas à trouver des régimes posologiques comparables pour tous les produits identifiés conformément au paragraphe 8.10 du chapitre 1 ou lorsque les prix de ces produits médicamenteux semblent excessifs. Exceptions
2.11 À moins que le breveté ne fasse valoir dans sa présentation que son produit médicamenteux offre de plus grands bienfaits thérapeutiques que les produits médicamenteux existants et que le GCMUH arrive à la conclusion que le nouveau produit médicamenteux breveté constitue une amélioration modeste ou importante :
- a) Lorsque le nouveau produit médicamenteux breveté est un produit mixte selon la définition qui en est donnée dans le paragraphe 8.13 du chapitre 1, son prix de lancement sera considéré excessif si son prix de transaction moyen national ou l’un de ses prix de transaction moyens du marché est plus élevé que la somme des prix des différentes substances qui le composent.
- b) Sous réserve de l’alinéa (c), le prix de lancement d’un nouveau produit médicamenteux breveté qui est une nouvelle présentation de la même entité chimique, dans une forme posologique identique ou comparable (appendice 2), un régime posologique identique ou comparable et la même indication ou utilisation sera considéré excessif si son prix de transaction moyen national ou l’un de ses prix de transaction moyens du marché est plus élevé que le prix obtenu avec le test de la relation raisonnable (appendice 4). Lorsque le régime posologique comparable est très différent, le prix de lancement du nouveau produit médicamenteux breveté sera considéré excessif si son prix de transaction moyen national ou l’un de ses prix de transaction moyens du marché est plus élevé que le plus élevé des prix des produits médicamenteux identifiés de la façon expliquée dans le paragraphe 8.12 du chapitre 1 aux fins d’une comparaison du prix selon la catégorie thérapeutique (appendice 3).
- c) Exceptions au test appliqué au prix décrit dans le paragraphe 2.11 (b) :
- (i) Le prix de lancement d’un nouveau produit médicamenteux générique breveté que Santé Canada a jugé bioéquivalent au produit médicamenteux de marque de référence vendu au Canada sera considéré excessif si son prix de transaction moyen national ou si l’un de ses prix de transaction moyens du marché est plus élevé que le prix du produit médicamenteux de marque de référence soumis au test de la relation raisonnable (appendice 4).
- (ii) le prix de lancement d’un nouveau produit médicamenteux générique breveté qui est une version produite sous licence d’un produit médicamenteux de marque breveté vendu au Canada sera considéré excessif si son prix de transaction moyen national ou si l’un de ses prix de transaction moyens du marché est plus élevé que le prix du produit médicamenteux de marque breveté soumis au test de la relation raisonnable (appendice 4).
Procédures :
2.12 À la lumière des résultats du test appliqué au prix des ventes de la première journée de vente du produit au Canada et aux données sur ses ventes, le personnel du Conseil communiquera au breveté une opinion provisoire à savoir si le prix de son nouveau produit médicamenteux breveté semble ou non excessif.
2.13 Le prix de lancement d’un nouveau produit médicamenteux breveté sera établi en calculant pour la période de lancement le prix de transaction moyen national du produit médicamenteux pour toutes les catégories de clients et pour toutes les provinces et tous les territoires ainsi que les prix de transaction moyens du marché pour les trois catégories de clients (hôpital, pharmacie et grossiste) et pour chaque province et chaque territoire (appendice 12).
2.14 La période de lancement est la période qui va de la date de la première vente au Canada du nouveau produit médicamenteux jusqu’à la fin du semestre de rapport (30 juin ou 31 décembre de chaque année) si cette période couvre plus d’un mois. Autrement, si la période entre la date de la première vente et la fin du semestre de rapport ne couvre pas un mois, la période de lancement ira de la date de la première vente à la fin du semestre de rapport suivant.
2.15 Le personnel du Conseil peut exclure de ses comparaisons tout produit médicamenteux breveté ou non breveté lorsqu’il a des motifs de croire que le prix de vente du produit est excessif.
2.16 Les prix de tous les produits médicamenteux essentiels utilisés dans les comparaisons seront évalués conformément aux tests de prix décrits dans les Lignes directrices.
3. Examen des prix des produits médicamenteux brevetés existants
3.1 Sous réserve des paragraphes 3.2 à 3.6, le prix d’un produit médicamenteux breveté existant sera considéré excessif lorsque :
- il est plus élevé que le taux de variation de l’indice des prix à la consommation (IPC) après application de la méthode de rajustement du prix selon l’IPC (appendice 9), ou
- il est plus élevé que le plus élevé des prix pratiqués dans les différents pays de comparaison (appendice 6).
3.2 Lorsque le prix de transaction moyen national ou le prix de transaction moyen d’un marché d’un produit médicamenteux augmente par rapport à l’exercice précédent par suite de la réduction ou de l’élimination d’un avantage et que le breveté démontre, preuves à l’appui, que l’augmentation du prix de son produit est exclusivement attribuable à la réduction ou à l’élimination des avantages, il peut dans un tel cas être approprié de rajuster les prix moyens non excessifs (prix national et prix du marché) à l’aide de la méthodologie de la BAISSE (appendice 10). Dans tous les cas, le prix au Canada d’un produit médicamenteux sera jugé excessif s’il dépasse le prix obtenu avec la comparaison du prix au Canada avec le prix international le plus élevé.
3.3 Par ailleurs, lorsque le breveté peut démontrer que l’augmentation du prix de transaction moyen national de son produit est exclusivement attribuable à un changement au niveau des ventes et non à une augmentation des prix de transaction moyens du marché pour chaque catégorie de clients et pour chaque province/territoire ne dépasse pas leurs prix de transaction moyens non excessifs calculés à l’aide de la méthodologie du rajustement du prix selon l’IPC, le prix de transaction moyen national ne sera alors pas considéré excessif.
3.4 Si le taux de variation de l’IPC est moins élevé que prévu et que le prix apparaît excessif du seul fait que le breveté a fixé le prix de son produit en fonction de l’IPC prévu, le prix du produit ne sera alors pas considéré excessif. Toutefois, pour les périodes de rapport subséquentes, le breveté devra rajuster le prix de son produit selon l’IPC réel. De même, l’application de la méthodologie de rajustement du prix selon l’IPC pour l’année de prévision se fondera sur le taux réel de variation de l’IPC de l’année. Quant aux brevetés qui ont augmenté les prix de leurs produits dans la mesure du taux d’inflation prévu, ce sera le taux réel de l’IPC de l’année de prévision qui sera utilisé pour calculer le prix moyen non excessif national et le prix moyen non excessif du marché.
3.5 Le Conseil est conscient que le breveté peut invoquer les coûts de réalisation et de mise en marché pour justifier un rajustement du prix moyen non excessif de son produit médicamenteux breveté (c.-à-d. lorsque l’Avis de conformité a été attribué au produit médicamenteux et que ce produit a été initialement vendu pour des motifs humanitaires à titre de nouvelle drogue de recherche, par le truchement d’une demande d’essai clinique ou, encore, au titre du Programme spécial d’accès).
3.6 Le CEPMB se réserve le droit de faire l’examen du prix de tout produit médicamenteux breveté existant disponible sur un marché au Canada (par ex. selon la catégorie de clients dans une province ou dans un territoire).
Procédures :
3.7 Le personnel du Conseil évaluera le prix de transaction moyen national par rapport au prix moyen non excessif national à l’aide de la méthodologie de rajustement du prix selon l’IPC et de la Comparaison du prix du produit médicamenteux au Canada avec son prix de vente le plus élevé dans les pays de comparaison.
3.8 Le CEPMB fera également un examen des variations des prix de transaction moyen sur différents marchés (hôpital, pharmacie, grossiste et chaque province et chaque territoire) dans les circonstances suivantes :
- lorsque le prix de transaction moyen national est plus élevé que le prix moyen non excessif national d’après les résultats obtenus avec la méthodologie du prix rajusté selon l’IPC et ce, dans une mesure justifiant la tenue d’une enquête (appendice 11 « Critères justifiant la tenue d’une enquête »)
- lorsque le CEPMB reçoit une plainte, ou
- Au besoin, dans le cadre d’une vérification de la conformité d’un engagement de conformité volontaire ou d’une ordonnance du Conseil.
3.9 Selon les résultats de l’enquête, le prix d’un produit médicamenteux breveté sera considéré excessif si son prix de transaction moyen national est plus élevé que son prix moyen non excessif national ou, encore, si les prix de transaction moyens du marché sont plus élevés que leurs prix moyen non excessifs respectifs. Vous trouverez dans l’appendice 12 la description du processus d’examen du prix du marché des produits médicamenteux brevetés existants.
4. Vente par un autre breveté d’un produit médicamenteux breveté existant
4.1 Lorsqu’un produit médicamenteux breveté existant est vendu au Canada par un breveté qui n’est pas le premier titulaire du brevet, le prix de transaction moyen national et le prix de transaction moyen du marché auxquels le nouveau breveté vend au départ son produit au Canada sera jugé excessif s’il est plus élevé que le prix moyen non excessif national et que les prix moyens non excessifs des marchés établis par la dernière période où le produit était vendu par l’ancien breveté.
4.2 Aux fins du calcul pour les périodes ultérieures du prix moyen non excessif national, décrit à l’appendice 9, le prix « de référence » pour le nouveau breveté sera le prix le moins élevé entre le prix moyen non excessif national de l’ancien détenteur du brevet et le prix de transaction moyen non excessif national du nouveau breveté.
4.3 Lorsque le nouveau breveté n’a pas accès aux prix de l’ancien breveté, c’est alors la méthodologie de rajustement du prix selon l’IPC qui doit être appliquée comme si le produit médicamenteux breveté en était à sa première année de vente sur le marché canadien.
4.4 Lorsque le prix de référence national du nouveau breveté correspond au prix moyen non excessif national de l’ancien breveté et que le nouveau breveté est en mesure de démontrer qu’il a eu accès au prix pratiqué par l’ancien breveté, la méthodologie de rajustement du prix selon l’IPC sera alors appliquée comme si le prix de vente du nouveau produit médicamenteux breveté se situait dans le prolongement du prix auquel l’ancien breveté vendait son produit médicamenteux. Comme le prévoit la Méthodologie de rajustement du prix selon l’IPC (appendice 9), le nouveau breveté sera alors autorisé à appliquer les variations du taux de l’IPC jusque-là non appliquées par l’ancien breveté.
5. Comparaison du prix selon la catégorie thérapeutique internationale
5.1 La comparaison du prix selon la catégorie thérapeutique internationale permet de comparer le prix du produit médicamenteux breveté avec les prix départ-usine des médicaments identifiés selon le test de la relation raisonnable ou de la comparaison de la classe thérapeutique, lesquels prix sont disponibles au grand public et pratiqués dans les pays de comparaison nommés dans le Règlement. La comparaison du prix selon la catégorie thérapeutique internationale ne sera effectuée que lorsqu’il semble qu’elle pourrait fournir des renseignements utiles en cas de différend à savoir si le prix du produit médicamenteux sous examen semble excessif à la lumière des résultats des autres tests (test de la relation raisonnable, comparaison selon la catégorie thérapeutique). Ce test, qui est décrit dans l’appendice 7, n’est pas utilisé comme test primaire.
Chapitre 3 – Enquêtes
1. Introduction
1.1 Lorsque le prix d’un produit médicamenteux breveté semble plus élevé que le prix autorisé en vertu des Lignes directrices, mais dans une mesure qui ne justifie pas une enquête (appendice 11), le breveté sera informé de la situation et mention sera faite dans le site Web du CEPMB que le prix du produit médicamenteux breveté semble excessif. En principe, le breveté devrait alors réduire le prix de transaction moyen national de son produit médicamenteux ainsi que les prix de transaction moyens du marché pour qu’ils ne soient pas excessifs et rembourser les recettes excessives qu’il a tirées de la vente de son produit à un prix excessif (appendice 13 – Remboursement des recettes excessives) mais le personnel du Conseil n’engagera dans l’immédiat aucune mesure pour recouvrer les recettes excessives.
1.2 Lorsque le prix de transaction moyen national d’un produit médicamenteux breveté semble plus élevé que le prix moyen non excessif national et que les circonstances justifient la tenue d’une enquête selon le critère établi par le Conseil (appendice 11), le breveté sera informé que le prix de son produit est sous enquête et que mention sera faite dans le site Web du CEPMB que le prix de son produit est « sous enquête ».
1.3 L’examen comportera une analyse de l’évolution du prix depuis le lancement du produit sur le marché canadien, de son prix de transaction moyen national et de ses prix de transaction moyen du marché et ce, pour chaque catégorie de clients (hôpital, pharmacie, grossiste) et de chaque province et de chaque territoire).
1.4 Habituellement, le breveté ne dispose que d’un bref délai pour donner suite à l’avis que lui a signifié le personnel du Conseil l’informant que le prix de son produit faisait l’objet d’une enquête. Par exemple, si le breveté aurait dû savoir à la lumière des rapports qu’il a soumis au Conseil (par ex. prix rajusté selon l’IPC) que le prix de son produit semblait plus élevé que ne l’autorisent les Lignes directrices (par ex. lorsque le prix a été majoré d’un taux plus élevé que celui autorisé avec la méthodologie du prix rajusté selon l’IPC), le personnel du Conseil lui accordera un délai aussi court que sept jours civils. Le délai peut aller jusqu’à 30 jours civils lorsqu’il semble que le breveté n’était pas conscient que le prix de transaction moyen national ou les prix de transaction moyens du marché de son produit médicamenteux étaient excessifs (par ex. lorsque le GCMUH a recommandé pour la comparaison de produits médicamenteux différents ou d’autres régimes posologiques que ceux proposés ou escomptés par le breveté).
1.5 L’enquête peut mener à l’un ou l’autre des trois résultats suivants :
- Le prix de transaction moyen national et (ou) les prix de transaction moyen du marché ne semblent pas excessifs
- Le prix de transaction moyen national et (ou) les prix de transaction moyens du marché semblent excessifs et le breveté soumet un Engagement de conformité volontaire satisfaisant aux exigences du Conseil ; ou
- Le prix de transaction moyen national et (ou) les prix de transaction moyens du marché semblent excessifs et le breveté n’a soumis aucun Engagement de conformité volontaire satisfaisant aux exigences du Conseil. Dans un tel cas, le personnel du Conseil soumet l’affaire à l’attention du président du Conseil et lui recommande d’émettre un Avis d’audience.
2. Prix qui ne semblent pas excessifs
2.1 Si l’enquête révèle que le prix de transaction moyen national et (ou) les prix de transaction moyens du marché ne semblent pas excessifs, l’enquête sera close et le breveté en sera informé.
3. Engagement de conformité volontaire
3.1 Si l’enquête confirme que le prix de transaction moyen national et (ou) les prix de transaction moyens du marché semblent excessifs, le breveté sera invité à soumettre un Engagement écrit de conformité volontaire prévoyant la réduction du prix de son produit médicamenteux et le remboursement des recettes excessives qu’il a tirées de la vente de son produit médicamenteux à un prix qui semble excessif (appendice 13).
3.2 L’Engagement de conformité volontaire que soumet le breveté ne constitue pas une admission de sa part que le prix de transaction moyen national et (ou) que les prix de transaction moyens du marché de son produit médicamenteux breveté sont ou ont été excessifs.
3.3 Le personnel du Conseil aidera le breveté à préparer son Engagement de conformité volontaire. À cette fin, il lui fournira un document type d’engagement de conformité volontaire ainsi que des conseils adaptés à sa situation.
3.4 Lorsque l’Engagement de conformité volontaire du breveté est jugé conforme aux Lignes directrices, le document d’engagement est soumis au président du Conseil (ou au panel d’audience lorsque l’Engagement de conformité volontaire est soumis au Conseil après l’émission de l’Avis d’audience) à qui il appartient de l’approuver s’il y a lieu.
3.5 Le président du Conseil n’est pas autorisé à négocier avec le breveté les modalités de son Engagement de conformité volontaire.
3.6 Dans l’Engagement de conformité volontaire, le breveté doit confirmer le prix moyen maximum potentiel de son produit médicamenteux pour la période de lancement, le prix de transaction moyen national et les prix moyens non excessifs du marché pour les autres périodes et s’engager à les respecter. Il doit également mentionner dans son Engagement la façon dont il remboursera les recettes excessives qu’il a tirées de la vente de son produit médicamenteux.
3.7 Dans la plupart des cas, l’Engagement de conformité volontaire contiendra une clause prévoyant le versement d’un montant à Sa majesté du chef du Canada aux fins de rembourser les recettes excessives tirées de la vente de son produit à un prix excessif.
3.8 Le président du Conseil (ou le Panel d’audience) prendra la décision d’accepter ou non l’Engagement de conformité volontaire à la lumière de l’article 83 de la Loi.
3.9 Le CEPMB fera rapport au public de tous les Engagements de conformité volontaire acceptés par le président du Conseil ou par un panel d’audience. Il fera nommément rapport du nom du produit médicamenteux breveté et (ou) du breveté et de tout autre élément d’information pertinent. Mention de ces éléments d’information sera également faite dans le rapport annuel ainsi que dans le site Web du CEPMB. Ces éléments d’information peuvent également être rapportés dans La Nouvelle ou dans d’autres publications.
Appendices
Ces appendices sont une partie intrégrante des Lignes directrices.
- Proposition du breveté sur le niveau d’amélioration thérapeutique
- Formes posologiques comparables
- Comparaison selon la catégorie thérapeutique
- Test de la relation raisonnable
- Test de la médiane des prix internationaux
- Comparaison du prix au Canada avec le prix international le plus élevé
- Comparaison du prix au Canada selon la catégorie thérapeutique internationale
- Tests appliqués aux prix des nouveaux produits médicamenteux
- Méthodologie de rajustement du prix selon l’IPC
- Méthodologie de la majoration
- Critères justifiant la tenue d’une enquête
- Examens du prix « sur un marché »
- Remboursement des recettes excessives
Appendice 1 – Proposition du breveté sur le niveau d’amélioration thérapeutique
Dans sa présentation, le breveté doit justifier sa proposition concernant le niveau d’amélioration des bienfaits thérapeutiques qui devrait être attribué à son produit médicamenteux, la sélection des produits médicamenteux pour les différentes comparaisons et les régimes posologiques comparables.
Le breveté doit soumettre sa proposition et ses documents de référence en dix copies papier. Le personnel du Conseil vérifiera l’ensemble de la proposition du breveté afin de s’assurer que tous les documents référence mentionnés ou listés dans le document ont été joints à la proposition. Advenant que certains documents soient manquants, le personnel du Conseil communiquera avec le breveté pour les obtenir.
1. Données cliniques à l’appui
1.1 Médicament : nom du médicament, sa catégorie, brève description de son mécanisme d’action, indications approuvées ou utilisations, dose approuvée ou proposée.
1.2 Monographie du produit (ou, si l’avis de conformité n’a pas encore été attribué au produit médicamenteux, les renseignements que contient normalement une monographie de produit) : présentée en même temps que le formulaire 1 « Renseignements identifiant le médicament ».
1.3 Essais individuels/études :
- Éléments de preuve de niveau 1 : Rapports publiés d’essais cliniques randomisés menés sur le produit médicamenteux sous examen versus les médicaments de comparaison actifs, s’il y en a; Rapports publiés d’essais cliniques randomisés portant sur le médicament sous examen versus un placébo; Rapports d’essais cliniques randomisés de grande qualité non publiés, lorsque disponibles.
- Rapports publiés d’essais cliniques utilisant des éléments de preuve de moindre niveau (par ex. études des résultats, examens systématiques des cohortes et des essais cas-témoins) lorsque les éléments de preuve de niveau 1 ne sont pas disponibles. Nota : En ce qui concerne les éléments de preuve de niveau 1 et d’autres niveaux, le breveté est encouragé à axer sa présentation sur les principaux essais cliniques qui ouvrent la voie à l’Avis de conformité ou à un changement de la pratique clinique ou qui pourraient constituer les meilleurs éléments de preuve du breveté.
- Éditoriaux et erratum se rapportant à des rapports d’essais cliniques publiés
- Autres éléments de preuve clinique, tels que études écologiques, cas et sondages sur le produit médicamenteux sous examen lorsque le breveté allègue que son produit apporte de plus grands bienfaits thérapeutiques associés aux facteurs secondaires.
1.4 Sommaire des essais mentionnés dans un tableau de la présentation :
- Études à consulter (Abrégés et publications publiés) et bibliographie fournie par le breveté.
- Brève description de l’étude ainsi que des indicateurs des résultats.
- Phase d’essai (Phase II, III ou IV); Les essais de phase I ne feront pas l’objet d’une révision.
1.5 Brève description des normes de thérapie ou de la pratique clinique acceptée pour lesquelles le produit médicamenteux sous examen est indiqué ou utilisé :
- Par exemple, examens de classe, analyses systématiques/méta-analyses.
1.6 Lignes directrices pour la pratique clinique publiées. Ces lignes directrices portent sur l’indication ou sur l’utilisation du produit médicamenteux sous examen si disponibles :
- de préférence, les lignes directrices canadiennes révisées par des pairs; les lignes directrices des États-Unis, du Royaume- Uni, de l’Australie et de l’Europe seront également prises en compte.
2. Proposition du breveté
2.1 Résumé :
- Brève description du produit médicamenteux et de la place qu’il occupe dans la thérapie ainsi que le sommaire des éléments de preuve clinique.
2.2 Niveau proposé sous l’angle de l’amélioration des bienfaits thérapeutiques
2.3 Produits médicamenteux proposés pour la comparaison :
- Éléments prouvant la même indication approuvée ou la même utilisation que le produit médicamenteux sous examen.
2.4 Régimes posologiques proposés pour la comparaison avec le produit médicamenteux sous examen :
- Doses approuvées ou proposées
- Doses utilisées dans les essais cliniques
- Doses recommandées dans les lignes directrices pour la pratique clinique.
3. Niveaux de preuve pour la détermination de l’amélioration des bienfaits thérapeutiques3
3.1 Le tableau qui suit présente la hiérarchie des éléments de preuve que considère le GCMUH au moment de formuler sa recommandation quant au niveau d’amélioration des bienfaits thérapeutiques que représente un nouveau produit médicamenteux breveté.
Niveau |
Thérapie/Prévention |
Analyses économiques et analyses des décisions |
1a |
RM (avec homogénéité*) d’ECR |
RM (avec homogénéité*) d’études économiques de niveau 1 |
1b |
ECR individuel (avec intervalle de confiance étroit) |
Analyse fondée sur des coûts ou des alternatives cliniquement sensibles; revue(s) méthodiques(s) des preuves; inclusion d’analyses de sensibilité multidirectionnelles |
1c |
Série de cas de type « tous ou aucun »§ |
Analyse de « meilleure valeur » ou de « pire valeur » absolue † |
2a |
RM (avec homogénéité*) d’études de cohortes |
RM (avec homogénéité*) d’études économiques de niveau >2 |
2b |
Étude de cohortes isolée (y compris ECR de faible qualité; par exemple avec un suivi de <80 %) |
Analyse basée sur des coûts ou des alternatives cliniquement sensibles : revue(s) limitée(s) des preuves ou études isolées; et inclusion d’analyses de sensibilité multidirectionnelles |
2c |
Études de « morbi-mortalité »; Études écologiques |
Audit ou études de « morbi-mortalité » |
3a |
RM (avec homogénéité*) d’études cas-témoins |
RM (avec homogénéité*) d’études de niveau 3b ou meilleures |
3b |
Étude cas-témoins individuelle |
Analyse fondée sur des alternatives ou des coûts limités, des estimations de mauvaise qualité des données, mais avec des analyses de sensibilité incorporant des variations cliniquement sensibles |
4 |
Série de cas (et études de cohortes et cas-témoins de mauvaise qualité §§) |
Analyse sans analyse de sensibilité |
5 |
Opinion d’expert sans évaluation critique explicite ou basée sur la physiologie, des travaux expérimentaux ou des « principes physiopathologiques primaires » |
Opinion d’expert sans évaluation critique explicite ou basée sur la physiologie des travaux expérimentaux ou des « principes physiopathologiques primaires » |
* Homogénéité désigne une revue méthodique sans variation préoccupante (hétérogénéité) des directions et des degrés des résultats des différentes études. Les revues méthodiques ayant une hétérogénéité statistique importante ne sont pas nécessairement préoccupantes et, inversement, les revues ayant une hétérogénéité préoccupante ne sont pas toutes statistiquement importantes. Tel que mentionné précédemment, les études ayant une hétérogénéité préoccupante doivent être identifiées au moyen de “-” suivi de leur niveau désigné.
§ Cas rencontrés lorsque tous les patients sont décédés avant que la thérapie ne soit disponible, mais certaines patients vivent encore grâce à ce médicament; d’autres sont décédés avant que la thérapie ne soit disponible. Aucun patient sous traitement ne décède.
§§ Une étude de cohorte de mauvaise qualité ne définit pas clairement les groupes de comparaison et (ou) ne mesure pas les expositions et la morbidité d’une façon objective (de préférence à l’aveugle) chez les patients exposés et non exposés et (ou) n’identifie pas ou ne contrôle pas d’une façon appropriée les variables confusionnelles connues.
† Traitements de meilleure valeur sont aussi bons, mais moins coûteux ou, encore, plus efficaces pour un même coût ou pour un coût moindre. Les traitements de pire valeur sont aussi bons et plus chers ou, encore, pires et aussi chers sinon plus chers.
RM : Revue systématique de la littérature
ECR : Essai clinique randomisé
Rx: Thérapie
Appendice 2 – Formes posologiques comparables
Le présent appendice présente les formes posologiques qui peuvent être utilisées pour le test de la relation raisonnable auquel sont soumis les nouveaux produits médicamenteux brevetés. Les formulations d’une même catégorie sont considérées comparables, mais les formes posologiques de catégories différentes ne le sont pas.
Le CEPMB vérifie de temps à autre la liste des formes posologiques comparables afin de s´assurer qu´elle contient toutes les formes posologiques utilisées.
| Formes posologiques comparables |
| Topique |
Nasale/Pulmonaire |
Voie orale (solide) |
| Aérosol |
Aérosol |
Caplet |
| Atomiseur |
Atomiseur |
Caplet à libération modifiée |
| Crème |
Gaz |
Comprimé |
| Disque |
Gouttes |
Comprimés à libération modifiée |
| Gel |
Poudre |
Gélule |
| Liquide |
Préparations en doses mesurées |
Gélules à libération modifiée |
| Onguent |
Solution |
Comprimés effervescents |
| Pansement |
|
Granules effervescentes |
| Pommade |
|
Poudre effervescente |
| Poudre |
|
|
| Shampoing |
|
|
| Voie orale (liquide) |
Vaginale |
Parentérale |
| Formulation à libération modifiée |
Comprimé |
Implant |
| Gouttes |
Cône |
Injection à libération modifiée |
| Poudre soluble |
Crème |
Poudre soluble |
| Poudre pour suspension |
Dispositif insérable |
Solution |
| Suspension |
Douche |
Suspension ou émulsion |
| |
Éponge |
|
| |
Gel |
|
| |
Mousse |
|
| |
Ovule |
|
| |
Suppositoire |
|
| |
Tampon |
|
| Otique/Opthalmique |
Rectale |
Dentaire/Sublinguale/Buccale |
| Dispositif oculaire à libération modifiée |
Crème |
Atomiseur sublingual |
| Gouttes |
Lavement |
Atomiseur buccal |
| Gel |
Mousse |
Comprimé buccal à libération modifiée |
| Liquide |
Onguent |
Comprimé sublingual |
| Onguent |
Suppositoire |
Gel |
| Poudre soluble |
Suspension |
Gomme |
| Suspension |
|
Pastille |
| |
|
Poudre pour suspension |
| |
|
Pâte dentifrice |
| |
|
Poudre dentifrice |
| |
|
Rince-bouche |
| |
|
Solution |
| |
|
Suspension |
Appendice 3 – Comparaison selon la catégorie thérapeutique
1. Approche
La comparaison selon la catégorie thérapeutique permet de comparer le prix de transaction moyen national et les prix de transaction moyens du marché pour chaque catégorie de clients – hôpital, pharmacie, grossiste et chaque province/territoire – avec les prix au Canada des produits médicamenteux retenus pour la comparaison que le CEPMB ne considère pas excessifs. Les prix des produits médicamenteux retenus pour la comparaison conformément èa la section 8 du chapitre 1, sont comparés avec ceux du nouveau produit médicamenteux breveté sous examen.
2. Mesure du prix
Le CEPMB considère qu’il est approprié de comparer les prix des produits médicamenteux utilisés pour les comparaisons de prix en tenant compte des régimes posologiques comparables déterminés conformément aux paragraphes 8.15 et 8.16 du chapitre 1. Selon le cas, le CEPMB effectuera les comparaisons de prix selon le coût/traitement ou le coût/jour. En règle générale, la comparaison sera faite selon le coût/traitement pour des maladies graves et selon le prix/jour (sur la base d’une dose d’entretien) pour les conditions chroniques.
Lorsque le produit médicamenteux utilisé pour la comparaison du prix est breveté et qu’il est vendu par le même breveté que celui qui vend le nouveau produit médicamenteux breveté, le prix du produit médicamenteux utilisé pour la comparaison correspondra à son prix de transaction moyen national.
Cependant, lorsque le produit médicamenteux utilisé pour la comparaison est breveté et qu’il est vendu par un autre breveté que celui qui a lancé le produit sous examen sur le marché canadien, le personnel du Conseil consultera les sources de prix publiques pour établir les prix des médicaments de comparaison. Le personnel du Conseil trouvera dans ces sources le prix public qui se rapproche le plus du prix moyen non excessif national du produit médicamenteux breveté utilisé pour la comparaison.
Lorsque le produit médicamenteux utilisé pour la comparaison n’est pas breveté, le personnel du Conseil consultera les sources de prix publiques pour s’assurer qu’il n’est pas vendu au Canada à un prix excessif. Le personnel du Conseil retiendra le prix public qui se rapproche le plus du prix moyen non excessif national établi par les Lignes directrices.
Le personnel du CEPMB peut exclure de sa comparaison de prix selon la catégorie thérapeutique tout produit médicamenteux lorsqu’il a des motifs de croire que ce produit est vendu à un prix excessif (voir la politique du CEPMB sur l’utilisation des produits médicamenteux brevetés et non brevetés dans les tests appliqués aux prix présentée dans la section 6 de la partie II – Politiques).
Appendice 4 – Test de la relation raisonnable
Pour que puisse être appliqué le test de la relation raisonnable, le nouveau produit médicamenteux breveté sous examen doit satisfaire les quatre exigences suivantes :
- Il doit avoir la même entité chimique que le produit médicamenteux retenu pour la comparaison
- Il doit avoir la même indication ou la même utilisation que le produit médicamenteux retenu pour la comparaison
- Il doit avoir la même forme posologique ou une forme posologique comparable que le produit médicamenteux retenu pour la comparaison (voir l’appendice 2), et
- Il doit également avoir le même régime posologique.
Le test de la relation raisonnable ne sera appliqué que si les quatre exigences susmentionnées sont satisfaites à moins que le breveté soumette une présentation dans laquelle il allègue que son produit offre de plus grands bienfaits thérapeutiques que les produits existants et que le Groupe consultatif sur les médicaments pour usage humain reconnaisse que le nouveau produit médicamenteux constitue une amélioration modeste ou importante des bienfaits thérapeutiques.
La relation raisonnable est le rapport entre la concentration de l’unité du produit médicamenteux (partie II – section 4) et son prix. Le test de la relation raisonnable définit le prix moyen maximal potentiel pour la nouvelle concentration du produit médicamenteux breveté.
Le présent appendice décrit en termes généraux le processus par lequel la relation raisonnable peut être vérifiée.
Si le produit médicamenteux de comparaison est breveté et qu’il est vendu par le breveté qui l’a lancé sur le marché canadien, le prix du produit médicamenteux utilisé pour la comparaison sera le prix de transaction moyen national de ce produit médicamenteux.
Si le produit médicamenteux de comparaison est breveté, mais qu’il est vendu par un autre breveté que celui qui a lancé le nouveau produit médicamenteux breveté sur le marché canadien, le personnel du Conseil consultera pour ces produits les sources de prix disponibles au grand public. Le personnel du Conseil trouvera dans ces sources des prix assez près du prix moyen non excessif national des produits médicamenteux brevetés utilisés pour la comparaison.
Lorsque le produit médicamenteux de comparaison n’est pas breveté, le personnel du Conseil utilisera pour ces produits les sources de prix disponibles au grand public si les prix obtenus ne sont pas excessifs. Le personnel du Conseil trouvera dans ces sources des prix assez près du prix moyen non excessif national déterminé par les Lignes directrices.
Le personnel du Conseil se réserve le droit d’exclure de ses tests sur la relation raisonnable tout produit médicamenteux s’il a des raisons de croire que le produit médicamenteux est vendu au Canada à un prix excessif (voir la politique du CEPMB sur l’utilisation des produits médicamenteux brevetés et non brevetés dans les tests appliqués au prix – partie II – Politiques, section 6).
La relation raisonnable peut être déterminée à l’aide d’un de trois tests possibles, mais un seul de ces tests peut être appliqué à un nouveau produit médicamenteux. Pour déterminer lequel des trois tests s’applique à un cas donné, il faut les considérer à tour de rôle en commençant par le test no 1 et en finissant par le no 3.
Test 1 : Lorsque les produits médicamenteux ont la même concentration
Lorsqu’un ou plusieurs produits médicamenteux retenus pour la comparaison du prix ont la même concentration que le nouveau produit médicamenteux breveté, c’est le produit médicamenteux de comparaison dont le prix est le plus élevé qui établit le prix moyen maximal potentiel du nouveau produit médicamenteux breveté. Tout prix supérieur au prix moyen maximal potentiel sera jugé excessif. Le résultat de ce test aura préséance sur ceux des deux autres tests sans égard aux résultats qu’ils produiront.
Dans le graphique 1 qui suit, étant donné que les trois produits médicamenteux retenus pour la comparaison ont la même concentration, mais des prix différents (P1, P2 et P3), le prix moyen maximal potentiel (PMMP) du nouveau produit médicamenteux breveté sera celui du produit médicamenteux dont le prix est le plus élevé soit, dans le présent cas, le produit P1.
Graphique 1 – Produits médicamenteux ayant la même concentration
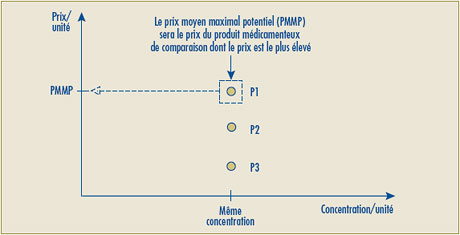
Test 2 : lorsque la relation du prix/unité est linéaire
Ce test est appliqué lorsqu’un minimum de deux produits médicamenteux sont retenus pour la comparaison de prix, mais qu’aucun de ces produits n’a la même concentration que le nouveau produit médicamenteux breveté.
Ce test comporte les étapes suivantes :
1. Comme on peut le voir dans le graphique 2A qui suit, des lignes sont tracées de manière à réunir autant de paires de produits médicamenteux que possible. Ces paires seront utilisées pour la comparaison (par ex. A à B, A à C et B à C).
2. C’est la paire créant une pente positive qui est égale à zéro ou plus élevée que zéro et dont le point d’intersection avec l’axe des ordonnées est le plus élevé qui détermine le prix moyen maximal potentiel. Dans le graphique 2A, le point d’intersection avec l’axe des ordonnées se situe dans la continuité de la ligne qui relie A à B.
Graphique 2A – Test de la relation linéaire – Étapes 1-2
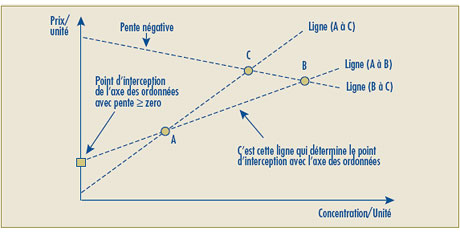
3. Une autre ligne est tracée de manière à joindre le point d’intersection sur l’axe des ordonnées avec le point représentant le prix/unité du produit médicamenteux de comparaison dont le prix est le plus élevé. Dans l’exemple du graphique 2B, c’est le produit médicamenteux C qui affiche le prix à l’unité le plus élevé.
4. Le prix moyen de transaction national et le prix moyen de transaction du marché du nouveau produit médicamenteux breveté ne seront pas excessifs s’ils ne dépassent pas la ligne correspondant au prix moyen maximal potentiel (PMMP) tracée dans le graphique 2B qui suit.
Graphique 2B – Test de la relation linéaire – Étapes 3-4
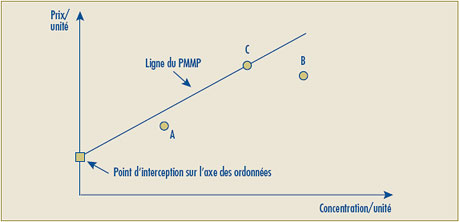
5. Il peut arriver qu’aucune des lignes réunissant une paire de produits médicamenteux de comparaison et dont la pente se situe à zéro ou plus que zéro ne croise l’axe des ordonnées au point zéro ou à un point plus élevé (lorsque tous les points d’intersection avec l’axe des ordonnées se situent sous zéro, le prix moyen maximal potentiel pour certaines concentrations sera également négatif). Dans un tel cas, la ligne du prix moyen maximal potentiel réunira le point d’origine (0) et le prix/unité du produit médicamenteux de comparaison dont le prix est le plus élevé. Dans l’exemple illustré dans le graphique 2C, le point d’origine sur l’axe des ordonnées serait négatif (si on avait utilisé la même méthodologie que pour l’étape 2). L’axe des ordonnées utilisé pour établir la ligne du prix moyen maximal potentiel ne pouvant se situer sous zéro, en conséquence le nouveau point d’intersection avec l’axe des ordonnées est établi au point d’origine, c’est-à-dire au point zéro (0). Dans cet exemple, la ligne du prix moyen maximal potentiel relie le point d’origine (0) au point du produit médicamenteux C. La ligne ainsi obtenue sera utilisée pour établir la relation entre la concentration du nouveau produit médicamenteux et son prix moyen maximal potentiel.
Graphique 2C – Test de la relation linéaire – Étape 5
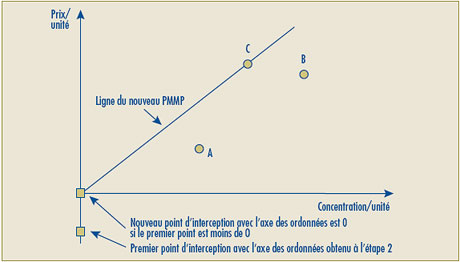
Test 3 : lorsque la concentration du produit médicamenteux utilisé pour la comparaison est différente de celle du nouveau produit médicamenteux breveté
Ce test est appliqué lorsque le produit médicamenteux utilisé pour la comparaison n’est vendu au Canada que dans une seule concentration et que cette concentration n’est pas la même (plus élevée ou moins élevée) que celle du nouveau produit médicamenteux sous examen. Dans ce test, c’est le produit médicamenteux de comparaison dont le prix est le plus élevé qui est retenu pour la comparaison lorsque les concentrations sont différentes.
Lorsque la concentration du nouveau produit médicamenteux breveté est plus élevée que celle du produit médicamenteux retenu pour la comparaison, le prix moyen maximal potentiel du nouveau produit médicamenteux breveté sera fonction de la relation proportionnelle de la concentration du nouveau produit médicamenteux breveté par rapport au produit médicamenteux de comparaison et sera multiplié par le prix du produit médicamenteux utilisé pour la comparaison.
Exemple 1 (lorsque la concentration du nouveau produit médicamenteux breveté est plus élevée) :
Le prix de vente le plus élevé auquel le produit médicamenteux de comparaison d’une concentration de 5 mg est 10 $.
Le nouveau produit médicamenteux breveté a une concentration de 7,5 mg.
Le prix du nouveau produit médicamenteux breveté de 7,5 mg sera considéré excessif s’il dépasse 15 $.
7,5 mg 5 mg X 10 $ = 15 $
Lorsque la concentration du nouveau produit médicamenteux breveté est moins grande que celle de son produit médicamenteux de comparaison, le prix moyen maximal potentiel du nouveau produit médicamenteux breveté sera le même que le prix du produit médicamenteux de comparaison présenté dans sa plus grande concentration.
Exemple 2 (lorsque le nouveau produit médicamenteux est présenté dans une concentration moins grande que son médicament de comparaison) :
Le prix de vente le plus élevé du produit de comparaison présenté dans une concentration de 5 mg est 10 $.
Le nouveau produit médicamenteux breveté est offert dans une concentration de 2,5 mg.
Ainsi, le prix de nouveau produit médicamenteux breveté présenté dans une concentration de 2,5 mg sera considéré excessif s’il dépasse 10 $.
Appendice 5 – Test de la médiane des prix internationaux
1. Comparaison du prix du produit médicamenteux au Canada avec la médiane de ses prix dans les pays de comparaison
1.1 C’est la médiane des prix départ-usine des mêmes concentrations et forme posologique du même produit médicamenteux breveté calculée pour chaque pays de comparaison nommé dans le Règlement (France, Allemagne, Italie, Suède, Suisse, Royaume-Uni et États-Unis) qui établit le prix moyen maximal potentiel d’un nouveau produit médicamenteux breveté lorsque le principal test appliqué au prix de lancement est la médiane des prix internationaux.
1.2 Lorsque le test appliqué au prix de lancement est la médiane des prix internationaux et que le nouveau produit médicamenteux breveté est vendu dans un nombre pair de pays, le prix médian ou la médiane correspondra généralement à la moyenne simple des deux prix du centre de la fourchette de prix.
1.3 Lorsque le nouveau produit médicamenteux breveté est vendu dans moins de cinq pays au moment de sa première vente du Canada, le prix international médian sera alors fixé sur une base intérimaire. Ainsi, après trois ans ou au moment où le même produit médicamenteux breveté (même concentration et même forme posologique) est vendu dans au moins cinq pays, soit la première de ces deux éventualités, le personnel du Conseil calculera à nouveau le prix international médian. Dans un tel cas, le prix moyen non excessif du produit médicamenteux sera le moindre des prix suivants :
- (a) le prix médian international révisé
- (b) le prix moyen non excessif obtenu avec la méthodologie de rajustement du prix selon l’IPC (appendice 9).
1.4 Lorsque le prix médian international révisé démontre, suivant l’application du paragraphe 1.3, que le prix moyen du produit médicamenteux est non excessif, le breveté devrait, de son propre chef, porter le prix de transaction moyen national et les prix de transaction moyens du marché au niveau du prix moyen non excessif calculé de la façon expliquée dans le paragraphe 1.3 et ce, avant la fin du deuxième semestre de rapport qui suit. Si le breveté réduit ainsi le prix de son produit médicamenteux dans le délai imparti, le prix de son produit ne sera pas réputé avoir été excessif.
2. Comparaison indirecte du prix international
2.1 Lorsqu’il n’est pas possible d’effectuer une comparaison directe du prix international du produit médicamenteux sous examen du fait que le produit médicamenteux n’est vendu qu’au Canada, la comparaison doit alors être faite à l’aide des concentrations les plus voisines des formes posologiques comparables (appendice 2) du même produit médicamenteux breveté.
3. Taux de change
3.1 Pour le calcul du prix international médian d’un nouveau produit médicamenteux breveté, les taux de change utilisés seront la moyenne simple de la moyenne des taux du cours au comptant à midi pour chaque pays (arrondie à la huitième décimale) publiés par la Banque du Canada pour la période de trente-six mois se terminant quatre mois avant la date de la première vente du produit médicamenteux au Canada. Par exemple, si la première vente du nouveau produit médicamenteux breveté sous examen a été effectuée en octobre 2008, les taux de change qui seront utilisés couvriront la période allant de juin 2005 à mai 2008.
3.2 Chaque mois, le CEPMB publie les taux de change dans son site Web sous la rubrique « Demandes courantes ».
Appendice 6 – Comparaison du prix au Canada avec le prix international le plus élevé
1. Comparaison du prix au Canada avec le prix international le plus élevé
1.1 Sous réserve du paragraphe 1.2, tant pour la période de lancement que pour les périodes de rapport subséquentes, le prix de transaction moyen national et les prix de transaction moyens du marché (pharmacie, hôpital, grossiste, provinces/territoires) seront considérés excessifs s’ils sont plus élevés que le plus élevé des prix de la même concentration et de la même forme posologique du même produit médicamenteux breveté pratiqués dans les différents pays de comparaison nommés dans le Règlement (ces pays sont la France, l’Allemagne, l’Italie, la Suisse, le Royaume-Uni et les États-Unis).
1.2 Lorsque le prix de transaction moyen national et les prix de transaction moyen du marché d’un produit médicamenteux breveté sont plus élevés au Canada que dans les pays de comparaison, le prix du produit au Canada ne sera pas considéré excessif s’il est égal ou moins élevé que le prix au Canada d’un produit médicamenteux breveté ayant une forme posologique identique ou comparable et une concentration identique ou comparable.
2. Taux de change
2.1 Pour la comparaison du prix au Canada avec le prix international le plus élevé, les taux de change utilisés seront la moyenne simple de la moyenne des taux du cours au comptant à midi pour chaque pays (arrondie à la huitième décimale) publiés par la Banque du Canada pour la période de trente-six mois se terminant quatre mois avant la date de la première vente du produit médicamenteux au Canada. Par exemple, si la première vente du nouveau produit médicamenteux breveté sous examen a été effectuée en octobre 2008, les taux de change qui seront utilisés couvriront la période allant de juin 2005 à mai 2008.
2.2 Pour le calcul du prix international le plus élevé d’un produit médicamenteux existant, les taux de change utilisés seront la moyenne simple de la moyenne des taux du cours au comptant à midi pour chaque pays (arrondie à la huitième décimale) publiés par la Banque du Canada pour la période de trente-six mois se terminant le dernier mois de la période d’établissement du prix sous examen. Par exemple, pour une période d’établissement du prix allant de juillet à décembre 2007, les taux de change utilisés seraient ceux des mois de janvier 2005 à décembre 2007.
2.3 Chaque mois, le CEPMB publie les taux de change dans son site Web sous la rubrique « Demandes courantes ».
3. Circonstances exceptionnelles visant les produits médicamenteux existants
3.1 Les Lignes directrices prévoient que les brevetés doivent prendre les mesures qui s’imposent lorsque les résultats d’une enquête démontrent que le prix d’un produit médicamenteux breveté semble excessif. Toutefois, certaines circonstances peuvent faire en sorte que le prix d’un produit médicamenteux breveté ne semble pas excessif au cours d’une période d’examen donnée, mais semble excessif la période suivante à la lumière des résultats de la Comparaison du prix au Canada avec le prix international le plus élevé. Les circonstances à l’origine de la situation peuvent fort bien échapper au contrôle du breveté. Voici trois exemples de telles circonstances :
- Variations du taux de change
- Organisme de réglementation d’un autre pays qui a forcé la réduction du prix du produit médicamenteux sur son territoire, ou
- Le produit médicamenteux qui était vendu au plus fort prix a été retiré du marché.
Lorsque l’une ou l’autre des circonstances susmentionnées se présente, le breveté sera informé que le prix de son produit médicamenteux breveté semble excessif et sera invité à réviser à la baisse le prix de transaction moyen national de son produit avant la fin des deux prochaines périodes de rapport, dans lequel cas, le prix de son produit ne sera pas réputé excessif. S’il ne baisse pas tel que demandé le prix de son produit dans les délais impartis, le breveté sera invité à soumettre au Conseil un Engagement de conformité volontaire et à rembourser les recettes excessives qu’il a tirées de la vente de son produit à un prix excessif. La valeur de ces recettes sera calculée rétroactivement à la première période de rapport où le prix était plus élevé que le prix obtenu avec la Comparaison du prix au Canada avec le prix international le plus élevé. Si le breveté choisit de ne pas soumettre au Conseil un Engagement de conformité volontaire, l’affaire sera alors portée à l’attention du Président aux fins qu’il émette un Avis d’audience.
Appendice 7 – Comparaison du prix au Canada selon la catégorie thérapeutique internationale
1. Concept et application
1.1 La comparaison du prix de vente du produit médicamenteux au Canada avec ses prix de vente dans les différents pays de comparaison (comparaison du prix au Canada selon la catégorie thérapeutique internationale) permet de comparer le prix du produit médicamenteux breveté sous examen avec les prix de vente des produits médicamenteux retenus pour la comparaison dans les sept pays de comparaison nommés dans le Règlement. Ces pays sont la France, l’Allemagne, l’Italie, la Suède, la Suisse, le Royaume-Uni et les États-Unis.
1.2 La comparaison du prix au Canada selon la catégorie thérapeutique internationale n’est pas considérée comme un test primaire. Toutefois, son application peut fournir des éléments d’information utiles dans le contexte d’une enquête sur le prix d’un produit médicamenteux qui semble excessif.
2. Sélection des produits médicamenteux aux fins de la comparaison
2.1 Aux fins de la comparaison du prix au Canada selon la catégorie thérapeutique internationale, les produits médicamenteux retenus pour la comparaison sont ceux utilisés pour la Comparaison du prix selon la catégorie thérapeutique. Vous trouverez dans la section 8 du chapitre 1 de plus amples renseignements sur la sélection des produits médicamenteux retenus pour la Comparaison du prix selon la catégorie thérapeutique.
2.2 En ce qui concerne les produits génériques utilisés pour la comparaison, seuls les produits qui sont vendus par le même breveté au Canada et dans le pays de comparaison sont retenus pour la comparaison. Autrement dit, si le produit médicamenteux générique utilisé pour la comparaison est vendu au Canada par la société « X », mais qu’il est vendu dans d’autres pays par les sociétés « X » et « Y », seul le produit médicamenteux générique vendu par la société « X » sera pris en compte dans la comparaison du prix au Canada selon la catégorie thérapeutique internationale.
3. Méthodes dérivées de la Comparaison du prix selon la catégorie thérapeutique internationale
3.1 Les deux méthodes suivantes peuvent être utilisées pour calculer le prix à l’aide de la comparaison du prix au Canada selon la catégorie thérapeutique internationale :
- Approche classique selon la catégorie : Les prix auxquels tous les produits médicamenteux utilisés dans les comparaisons sont vendus dans les sept pays de comparaison nommés dans le Règlement sur les médicaments brevetés sont établis. Le prix international médian est calculé et comparés aux prix de transaction moyen national du produit médicamenteux breveté au Canada.
- Approche du ratio : Les prix du produit médicamenteux sous examen dans les sept pays de comparaison nommés dans le Règlement sont déterminés. Les prix auxquels tous les produits médicamenteux retenus pour la comparaison sont vendus dans les sept pays de comparaison sont également déterminés. Les ratios du prix du produit médicamenteux sous examen par rapport aux prix des produits médicamenteux utilisés dans la comparaison sont calculés pour chaque combinaison de produits dans chaque pays de comparaison. La médiane de tous les ratios ainsi obtenus est par la suite appliquée au prix de transaction moyen national du produit médicamenteux breveté sous examen au Canada.
3.2 Lorsqu’un produit médicamenteux qui devait être utilisé pour la comparaison a été exclu de la Comparaison du prix selon la catégorie thérapeutique du fait qu’il semblait excessif, ce produit est également exclu de la Comparaison du prix selon la catégorie thérapeutique internationale.
4. Taux de change
4.1 Pour la comparaison du prix d’un nouveau produit médicamenteux breveté selon la catégorie thérapeutique internationale, les taux de change utilisés correspondent à la moyenne simple de la moyenne des taux du cours au comptant à midi pour chaque pays (arrondie à la huitième décimale) publiés par la Banque du Canada pour la période de trente-six mois se terminant quatre mois avant la date de la première vente du produit médicamenteux au Canada. Par exemple, si la première vente du nouveau produit médicamenteux breveté sous examen a été effectuée en octobre 2008, les taux de change qui seront utilisés pour la comparaison couvriront la période allant de juin 2005 à mai 2008.
4.2 Pour la comparaison du prix d’un produit médicamenteux breveté existant selon la catégorie thérapeutique internationale, les taux de change utilisés correspondent à la moyenne simple de la moyenne des taux du cours au comptant à midi pour chaque pays (arrondie à la huitième décimale) publiés par la Banque du Canada pour la période de trente-six mois se terminant le dernier mois de la période d’établissement du prix du produit médicamenteux sous examen. Par exemple, pour une période d’établissement du prix qui irait de juillet à décembre 2007, les taux de change utilisés seraient ceux des mois de janvier 2005 à décembre 2007.
4.3 Chaque mois, le CEPMB publie les taux de change dans son site Web sous la rubrique « Demandes courantes ».
Appendice 8 – Tests appliqués aux prix des nouveaux produits médicamenteux
| Niveau d’amélioration des bienfaits thérapeutiques |
Test appliqué au prix d’un nouveau produit médicamenteux breveté |
| Découverte |
Test de la médiane des prix internationaux |
| Amélioration importante |
Le prix le plus élevé entre :
1) Le plus élevé des prix obtenus avec la Comparaison du prix selon la catégorie thérapeutique à laquelle ont été soumis tous les produits médicamenteux identifiés par le GCMUH en application du paragraphe 8.6 du chapitre 1; et
2) La médiane des prix internationaux |
| Amélioration modeste |
Le plus élevé des prix entre :
1) la médiane de : i) des prix obtenus avec la Comparaison du prix selon la catégorie thérapeutique de tous les produits médicamenteux identifiés par le GCMUH en application du paragraphe 8.7 du chapitre 1 et ii) la médiane des prix internationaux; et
2) prix le plus élevé obtenu avec la Comparaison du prix selon la catégorie thérapeutique de tous les produits médicamenteux identifiés par le GCMUH en application du paragraphe 8.7 du chapitre 1. Lorsqu’il n’est pas possible d’effectuer la comparaison du prix selon la catégorie thérapeutique (c.-à-d. lorsqu’on ne peut établir un régime posologique comparable ou lorsque le prix du seul produit médicamenteux qui se prête à la comparaison est excessif), utiliser alors la médiane des prix internationaux. |
| Amélioration minime ou nulle |
1) Le prix le plus élevé obtenu avec la comparaison du prix selon la catégorie thérapeutique de tous les produits médicamenteux comparables identifiés par le GCMUH en application du paragraphe 8.9 du chapitre 1.
2) Dans les cas exceptionnels, lorsque le GCMUH n’a réussi à identifier aucun produit médicamenteux comparable, utiliser alors le prix le moins élevé obtenu avec la comparaison du prix selon la catégorie thérapeutique des produits médicamenteux de la gamme supérieure identifiés par le GCMUH en application du paragraphe 8.10 du chapitre 1 et ii) la médiane des prix obtenus avec la médiane des prix internationaux.
3) Lorsqu’il n’est pas possible d’effectuer la comparaison du prix selon la catégorie thérapeutique (c.-à-d. lorsqu’on ne peut établir un régime posologique comparable ou lorsque le prix du seul produit médicamenteux qui se prête à la comparaison semble excessif), utiliser alors la médiane des prix internationaux.
Veuillez noter que les produits médicamenteux sousmentionnés sont assujettis aux Lignes directrices additionnelles à moins que le breveté n’allègue dans sa présentation que son produit offre de plus grands bienfaits thérapeutiques que les médicaments existants et que le GCMUH confirme que le nouveau produit médicamenteux constitue une amélioration modeste ou importante par rapport aux produits médicamenteux existants :
- produit médicamenteux combiné
- produit médicamenteux générique breveté
- produit médicamenteux qui est une nouvelle présentation de la même entité chimique avec une forme posologique identique ou comparable (Appendice 2), le même régime posologique et la même indication ou utilisation.
Les Lignes directrices qui s’appliquent à ces produits médicamenteux sont présentées dans le paragraphe 2.11 du chapitre 2. |
Appendice 9 – Méthodologie de rajustement du prix selon l’IPC
1. Méthodologie de rajustement du prix selon l’indice des prix à la consommation (IPC)
1.1 Sous réserve du paragraphe 1.5 et l’appendice 10, le prix de transaction moyen national et les prix de transaction moyens du marché d’un produit médicamenteux breveté existant seront considérés excessifs lorsque leur taux d’augmentation est plus grand que celui autorisé en vertu de la méthodologie de rajustement du prix selon l’IPC.
1.2 La méthodologie de rajustement du prix selon l’indice des prix à la consommation prévoit les calculs suivants :
- réviser les prix de référence du produit médicamenteux pour tenir compte des variations cumulatives de l’IPC entre l’année de référence et l’année sous examen (Prix rajusté selon l’IPC) ; et
- imposer une limite annuelle à l’augmentation du prix maximal (1,5 fois le taux annuel prévu de variation pour l’année de l’indice des prix à la consommation). Dans les périodes de grande inflation (plus de 10 %), la limite sera de 5 % de plus que le taux prévu de variation de l’IPC.
1.3 C’est le résultat le plus bas des deux calculs qui établit le prix moyen non excessif pour l’année.
1.4 Un calcul du prix moyen non excessif sera effectué pour chaque marché (marché national, catégorie de clients (pharmacie, hôpital, grossiste), chaque province et territoire) en tenant compte de l’historique du prix sur le marché.
1.5 L’exception qui suit à la méthodologie du prix rajusté selon l’IPC s’applique lorsque les recettes excessives sont remboursées en portant le prix du produit médicamenteux sous le prix moyen non excessif sur un ou sur plusieurs marchés (appendice 13 – Remboursement des recettes excessives). Lorsque les recettes excessives auront été remboursées, le breveté peut à la prochaine période de rapport augmenter les prix de son produit jusqu’à concurrence des prix de transaction moyens non excessifs du marché qui étaient pratiqués avant la réduction du prix.
2. Terminologie
2.1 Période de prévision :
Année pour laquelle les prix sont établis.
2.2 Période de lancement :
Dans le cas des nouveaux produits médicamenteux brevetés, la période de lancement est la période qui s’écoule entre la date de la première vente du produit médicamenteux et la fin du semestre de rapport (soit le 30 juin et le 31 décembre de chaque année) lorsque cette période s’étend sur plus d’un mois. Par exemple, la période de lancement d’un produit médicamenteux breveté vendu pour la première fois en mars 2007 irait de mars à juin 2007. Dans le cas d’un produit médicamenteux breveté lancé sur le marché canadien quelque temps en décembre 2007, sa période de lancement irait de janvier à juin 2008.
2.3 Année de référence :
- Dans le cas de produits médicamenteux brevetés lancés sur le marché canadien plus de trois ans avant la période de prévision, l’année de référence sera la troisième année précédant la période de référence. Par exemple, pour 2009, l’année de référence serait 2006.
- Dans le cas des produits médicamenteux brevetés lancés sur le marché au plus trois ans avant la période de prévision, l’année de référence sera l’année au cours de laquelle a été faite la première vente du produit au Canada.
2.4 Prix de référence :
- Dans le cas d’un produit médicamenteux breveté dont la première vente au Canada remonte à plus de trois ans avant la période de prévision, ses prix de référence national et du marché du produit médicamenteux breveté seront respectivement son prix de transaction moyen national et ses prix de transaction moyens du marché pour l’année de référence fondés sur le formulaire 2 du breveté, section 3, ou, si ces prix semblent excessifs, le prix moyen non excessif national et les prix moyens non excessifs du marché de l’année de référence.
- Dans le cas d’un produit médicamenteux breveté dont la première vente remonte à trois ans ou moins avant la période de prévision, les prix de référence national et du marché du produit seront respectivement son prix de transaction moyen national et ses prix de transaction moyens du marché de la période de rapport, fondés sur le formulaire 2 du breveté, section 4, ou, si les prix semblent excessifs, le prix moyen non excessif national et les prix moyens non excessifs du marché pour la période de lancement.
2.5 IPC de base :
Moyenne annuelle des augmentations mensuelles de l’IPC publiée par Statistique Canada pour l’année de référence. Chaque année, le CEPMB publie l’IPC de base dans la livraison d’avril de La Nouvelle.
2.6 IPC prévu :
IPC réel de l’année précédente publié par Statistique Canada et rajusté en fonction des plus récentes prévisions du taux annuel d’inflation publiées par le ministère fédéral des finances. Le CEPMB publie aussi chaque année l’IPC prévu dans la livraison d’avril de La Nouvelle.
2.7 Facteur de rajustement selon l’IPC :
L’IPC prévu est divisé par l’IPC de base et arrondi à la troisième décimale.
2.8 Prix rajusté selon l’IPC :
Prix de référence multiplié par le facteur de rajustement selon l’IPC pour l’année de référence.
2.9 Plafond :
Pour toute année, le prix d’un produit médicamenteux breveté ne peut augmenter de plus de 1,5 fois le taux prévu de variation de l’IPC de l’année. En période de forte inflation (plus de 10%), l’augmentation du prix du produit médicamenteux ne pourra être plus grande que l’IPC prévu plus 5 %.
2.10 Voici comment est appliquée la méthodologie du prix rajusté selon l’IPC :
Période de prévision : Janvier à décembre 2009
Année de la première vente : 1998
Année de référence : 2006
Prix de transaction moyen national pour l’année de référence : 10,00 $
Prix de transaction moyen national en 2008 : 10,39 $
Prix rajusté selon l’IPC : 1,065 (facteur de rajustement selon l’IPC pour 2006) × 10,00 $ = 10,65 $
Plafond : 1,030 (1,5 x IPC prévu pour 2009 qui est dans le présent exemple 2,0 %) × 10,39 $ = 10,70 $
Ainsi, le prix moyen non excessif national pour 2009 est 10,65 $.
Appendice 10 – Méthodologie de la majoration
1. Méthodologie de la majoration
1.1 Définition
Lorsque le breveté soutient que le prix de son produit médicamenteux a augmenté dans une mesure plus grande que ne le permet la méthodologie du prix rajusté selon l’IPC parce qu’il a réduit ou cessé d’offrir des avantages à ses clients et qu’il présente des éléments de preuve à l’appui, le prix moyen non excessif national du produit et (ou) le prix moyen non excessif du marché pourront augmenter dans une mesure plus grande que celle autorisée en vertu de la méthodologie de rajustement du prix selon l’IPC.
1.2 Avantages admissibles
Les avantages sont, par définition, « toute réduction accordée à titre de promotion ou sous forme de rabais, d’escompte, de remboursement, de biens ou de services gratuits, de cadeaux ou de tout autre avantage de semblable nature ».
1.3 Éléments de preuve requis
Les brevetés qui souhaitent invoquer la méthodologie de la majoration doivent présenter les éléments suivants :
- Élément démontrant que, avant même de bénéficier de l’avantage, les clients étaient conscients que l’avantage qui leur était offert n’était pas offert à tous les clients
- Le type et la valeur de l’avantage, le moment qu’il a été offert ainsi que la façon dont il a été offert
- Éléments prouvant la réduction ou la cessation de l’avantage
- Déclaration à savoir si le même client reçoit encore ou non d’autres avantages.
La forme exacte de ces éléments de preuve (par ex. contrat) et le type de données (par ex. quantité du produit offert gratuitement, escompte, rabais) varieront selon le cas.
La méthodologie de la majoration ne s’appliquera pas aux cas où l’augmentation du prix de transaction moyen national semble n’être attribuable qu’à une modification de l’ensemble des ventes. Elle ne s’appliquera pas non plus lorsque le prix a été réduit pour rembourser les recettes excessives (appendice 13).
1.4 Application de la méthodologie de la majoration au calcul des prix moyens non excessifs
Lorsque le prix de transaction moyen national et les prix de transaction moyens du marché d’un produit médicamenteux augmentent dans une mesure plus grande que celle autorisée avec la méthodologie de rajustement du prix selon l’IPC et que le breveté présente les éléments de preuve exigés, le prix maximal auquel le produit peut être vendu sans être considéré excessif sera le suivant :
- Lorsque l’avantage a été offert après le lancement du produit sur le marché canadien, les prix de transaction moyens du marché pourront augmenter jusqu’à concurrence de son prix de transaction moyen antérieur le plus élevé sur ce marché, sans que ces prix ne semblent excessifs
- Lorsque l’avantage a été offert dès le moment où le produit a été lancé sur le marché et par la suite pendant chaque période de rapport, les prix de transaction moyens du marché où a été offert l’avantage peuvent augmenter jusqu’à concurrence du prix de transaction moyen le plus élevé sur tout marché comparable où aucun avantage n’a été offert sans que ces prix ne soient jugés excessifs. On entend par marché comparable le marché d’un hôpital, d’un grossiste et d’une pharmacie ainsi que chaque province et chaque territoire.
1.5 Exemple de l’application de la méthodologie de la majoration
| |
Hôpital PTMM* |
Grossiste PTMM* |
Pharmacie PTMM* |
PTM** national |
| Année 1 (Lancement) |
8,00 $ |
9,00 $ |
10,00 $ |
9,00 $ |
| Année 2 |
6,00 $ |
8,00 $ |
10,00 $ |
8,00 $ |
| Année 3 |
10,00 $ |
9,00 $ |
10,00 $ |
9,67 $ |
* PTMM veut dire Prix de transaction moyen du marché
** PTM veut dire Prix de transaction moyen national
L’année 3, le prix de transaction moyen national augmente au-delà de la mesure généralement permise par la méthodologie de rajustement du prix selon l’IPC. Le breveté formule les allégations suivantes avec éléments de preuve à l’appui :
i) Hôpital :
- L’année 1, à la date de la première vente, des contrats consentant un escompte de 20 % du prix de liste ont été négociés avec des organismes de groupement d’achats.
- L’année 2, en raison de leurs volumes élevés d’achats, certains hôpitaux ont pu bénéficier d’escomptes plus importants.
- L’année 3, un nouveau compétiteur a investi le marché des hôpitaux et les contrats existants n’ont pas été reconduits. Les hôpitaux qui ont continué d’acheter le produit médicamenteux du breveté ont payé le plein prix, soit 10,00 $. Ce prix est jugé non excessif à la lumière des éléments de preuve fournis.
ii) Grossiste :
- La première année, les grossistes ont payé le produit médicamenteux 9,00 $ puis, l’année 2, un rabais temporaire a été consenti à certains clients. À l’année 3, le rabais n’était plus offert.
- Le breveté a présenté des éléments de preuve à l’appui du prix préférentiel accordé l’année 2. Aucun élément de preuve n’a été présenté à l’appui d’un avantage consenti l’année où le produit a été lancé sur le marché. Ainsi, le prix de 9,00 $ n’est pas considéré excessif, des éléments de preuve ayant été fournis pour confirmer l’avantage.
Appendice 11 – Critères justifiant la tenue d’une enquête
En vertu de ses Lignes directrices, le Conseil peut se doter de critères pour l’aider à reconnaître les cas où il y a lieu de tenir une enquête sur le prix d’un produit médicamenteux qui semble excessif.
Ces critères établissent un juste équilibre entre la latitude accordée aux brevetés au niveau de l’établissement des prix de leurs produits médicamenteux et le mandat du CEPMB de veiller à ce que les produits médicamenteux brevetés ne soient pas vendus au Canada à des prix excessifs. Le Conseil publie les critères qui justifient la tenue d’une enquête sur le prix d’un produit médicamenteux breveté afin d’améliorer la transparence de son processus d’examen du prix et, également, de mieux sensibiliser les brevetés à leurs responsabilités en matière de conformité au Règlement.
Le prix de lancement d’un produit médicamenteux breveté est habituellement considéré non excessif lorsque son prix de transaction moyen national et ses prix de transaction moyens du marché sont égaux ou moins élevés que le prix maximal moyen potentiel. Pour les autres périodes de rapport, ces prix doivent être moins élevés que leurs prix moyens non excessifs.
Pour optimiser l’utilisation qu’il fait des ressources qu’il affecte aux enquêtes, le Conseil a élaboré des critères qui déterminent les circonstances où il y a lieu de tenir une enquête lorsque le prix d’un produit médicamenteux est plus élevé que ne l’autorisent les Lignes directrices.
| Critères qui justifient la tenue d’une enquête sur le prix |
Le personnel du Conseil entreprend une enquête sur le prix d´un produit médicamenteux breveté lorsqu’un des critères suivants s’applique :
- Le prix de transaction moyen national ou le prix de transaction moyen du marché d’un nouveau produit médicamenteux breveté dépasse de plus de 5 % le prix moyen maximal potentiel pour la période de lancement.
- Le prix de transaction moyen national d’un produit médicamenteux breveté existant dépasse de plus de 5 % le prix moyen non excessif national.
- Dans le cas d’un produit médicamenteux nouveau ou existant, la valeur des recettes excessives totalise 50 000 $ ou plus.
- Le CEPMB reçoit une plainte selon laquelle le prix du produit médicamenteux est excessif.
|
Lorsque, pour une année donnée, le prix de transaction moyen national dépasse le prix moyen non excessif national dans une mesure trop modeste pour justifier la tenue d’une enquête, le breveté sera invité à réduire le prix de son produit médicamenteux breveté pour qu’il se situe sous le prix moyen non excessif national ainsi qu’à rembourser les recettes excessives de la façon dont le prévoit la politique du CEPMB sur le remboursement des recettes excessives (Partie II – Politiques, section 7). Lorsqu’il est démontré que le breveté a pratiqué des prix supérieurs au prix moyen maximal potentiel durant la période de lancement ou au prix moyen non excessif national durant les périodes de rapport suivantes, ne serait-ce que de montants qui ne justifient pas la tenue d’une enquête, le personnel du CEPMB peut décider de tenir une enquête sur le prix.
Si le prix de transaction moyen national d’un produit médicamenteux breveté ou si la valeur cumulative des recettes excessives tirées de la vente du produit à un prix excessif est suffisamment élevée pour justifier la tenue d’une enquête, le personnel du Conseil engagera une enquête en application des Lignes directrices. Les brevetés seront informés du statut du prix de leur produit médicamenteux ainsi que de la valeur des recettes excessives, ventilées par produit médicamenteux, dans le cadre de rapports de conformité que leur fera parvenir le CEPMB.
Appendice 12 – Examens du prix « sur un marché »
En ce qui concerne les nouveaux produits médicamenteux brevetés
- Le personnel du Conseil appliquera le test approprié au prix de lancement du produit. Le choix du test variera selon le niveau d’amélioration des bienfaits thérapeutiques évalué. Le test déterminera le prix moyen maximal potentiel.
- Le prix moyen maximal potentiel sera appliqué au prix de lancement du produit sur chaque marché – c.-à-d. prix national, chaque catégorie de clients (hôpital, pharmacie et grossiste) et à chaque province/territoire.
Exemple 1
| Prix moyen maximal potentiel = |
10 $ |
|
Prix ne sera pas considéré excessif étant donné que le prix de transaction moyen national et les prix de transaction moyens du marché ne sont pas plus élevés que le prix moyen maximal potentiel. |
| PTM* national = |
9 $ |
 |
| PTM* du marché (Hôpital) = |
8 $ |
|
| PTM* du marché (Grossiste) = |
9 $ |
|
| PTM* du marché (Pharmacie) = |
10 $ |
|
* PTM signifie Prix de transaction moyen
Exemple 2
| Prix moyen maximal potentiel = |
10 $ |
|
Même si le prix de transaction moyen national n’est pas considéré excessif, le prix de transaction moyen du marché (pharmacie) est plus élevé que le prix moyen maximal potentiel et de ce fait sera considéré excessif. |
| PTM* national = |
9 $ |
|
| PTM* du marché (Hôpital) = |
6 $ |
|
| PTM* du marché (Grossiste) = |
9 $ |
|
| PTM* du marché (Pharmacie) = |
12 $ |
X |
* PTM signifie Prix de transaction moyen
Produits médicamenteux brevetés existants
- Après la période de lancement, le personnel du Conseil ne fera l’examen que du prix de transaction moyen national du produit médicamenteux breveté et le comparera au prix moyen non excessif national en utilisant la méthodologie du prix rajusté selon l’IPC et la Comparaison du prix avec le prix international le plus élevé.
- Le personnel du Conseil fera aussi enquête sur les variations des prix au niveau des marchés (catégorie de clients et (ou) province/territoire) lorsque :
- le prix de transaction moyen national est plus élevé que le prix moyen non excessif national dans une mesure qui justifie la tenue d’une enquête (appendice 11)
- le CEPMB reçoit une plainte, ou,
- cela est demandé aux fins d’un suivi de la conformité à un engagement de conformité volontaire ou à une ordonnance du Conseil.
- Lorsque le prix de transaction moyen national justifie la tenue d’une enquête, le personnel du Conseil analysera les tendances des prix sur chaque marché (par ex. hôpital, pharmacie, grossiste et chaque province/territoire) afin d’identifier quels marchés sont à l’origine d’un prix de transaction moyen national qui semble excessif.
- À cette fin, chaque prix de transaction moyen du marché sera comparé à chaque prix moyen non excessif du même marché après application de la méthodologie du prix rajusté selon l’IPC et de la comparaison du prix au Canada du produit médicamenteux avec le prix le plus élevé dans les pays de comparaison.
- Un des trois scénarios suivants peut se produire :
- Aucun prix de transaction moyen du marché n’est considéré excessif. Ce scénario peut se produire lorsque l’augmentation apparente du prix de transaction moyen national est exclusivement attribuable à un changement au niveau de la composition des ventes – c.-à-d. changement du volume des ventes sur chaque marché avec un plus grand volume vendu dans un marché dont le prix de transaction moyen du marché est plus élevé que dans la période de rapport précédente.
- Les prix d’au moins un marché ont été majorés et ainsi portés à un niveau qui semble excessif. En conséquence, le breveté devra baisser le prix de transaction moyen du marché au niveau du prix moyen non excessif pour le marché pertinent. Après cette réduction, le prix de transaction moyen national sera généralement considéré non excessif. Plutôt que de calculer les recettes excessives en se fondant exclusivement sur le(s) marché(s) où le prix du produit a été jugé excessif, les recettes excessives seront calculées sous l’angle du montant généré au niveau du prix de transaction moyen national.
- Le breveté peut soumettre des éléments de preuve pour démontrer que l’augmentation du prix sur le marché en cause était exclusivement attribuable au fait qu’il a réduit l’avantage offert sur ce marché ou qu’il y a mis fin. Dans un tel cas, il peut être approprié de dévier de la méthodologie du prix rajusté selon l’IPC pour utiliser plutôt la méthodologie de la majoration présentée dans l’appendice 10.
Appendice 13 – Remboursement des recettes excessives
Mesures de remboursement des recettes excessives
1.1 En vertu du paragraphe 1.3.1, lorsque la valeur des recettes excessives n’est pas suffisante pour justifier la tenue d’une enquête, le breveté sera invité à appliquer une réduction volontaire du prix de son produit médicamenteux afin de rembourser les recettes excessives qu’il a encaissées.
1.2 Par contre, si la valeur des recettes excessives est suffisamment élevée pour justifier la tenue d’une enquête, le breveté pourra rembourser les recettes excessives qu’il a encaissées suivant les modalités prévues dans un Engagement de conformité volontaire approuvé ou selon une décision du Conseil.
Délai de remboursement des recettes excessives
1.3 Les brevetés doivent rembourser avec diligence les recettes excessives qu’ils ont encaissées. À cette fin, les paramètres suivants seront généralement appliqués au moment de déterminer les modalités du remboursement des recettes excessives :
- 1.3.1 Les recettes excessives dont la valeur n’est pas suffisante pour justifier la tenue d’une enquête, mais qui ont été encaissées durant six semestres consécutifs (3 ans) devront être remboursées suivant les modalités convenues dans un Engagement de conformité volontaire. À défaut du breveté de négocier un Engagement de conformité volontaire, le personnel soumettra l’affaire à l’attention du président du Conseil.
- 1.3.2 Dans le contexte d’un Engagement de conformité volontaire et sous réserve des modalités de l’Engagement, le breveté devra rembourser les recettes excessives qu’il a encaissées dans un des délais suivants :
- dans les trente jours qui suivront l’acceptation de l’Engagement de conformité volontaire par le président du Conseil, ou
- d’ici à la fin de la prochaine période de rapport lorsque les recettes excessives sont remboursées au moyen d’une réduction du prix. Le reliquat des recettes excessives qui n’auront alors pas été remboursées à la fin de cette période de rapport deviendront exigibles.
Retour au prix normal après le remboursement complet des recettes excessives au moyen d’une réduction du prix
1.4 L’exception suivante à la méthodologie de rajustement du prix selon l’IPC s’applique lorsque le prix du produit a été réduit sous le prix moyen non excessif sur un ou plusieurs marchés aux fins de rembourser les recettes excessives. Ainsi, lorsque les recettes excessives auront été complètement remboursées, les prix de transaction moyens sur ces marchés pourront être rétablis dès la prochaine période de rapport jusqu’à concurrence des prix moyens non excessifs du marché qui avaient cours avant que ne soit appliquée la réduction du prix.
1 ICN Pharmaceuticals Inc. c. Canada (personnel du Conseil d’examen du prix des médicaments brevetés (C.A.) [1997] 1 C.F. 32, conf., jugement no 206 (T.D.)
2 Supra 1
3 Le tableau présenté est celui du Oxford Centre for Evidence-based Medicine Levels of Evidence (Mai 2001) – Il est élaboré depuis novembre 1998 par Bob Phillips, Chris Ball, Dave Sackett, Doug Badenoch, Sharon Straus, Brian Haynes et Martin Dawes.